使用R和Stata软件实现倾向性评分匹配
2018-09-20南通大学公共卫生学院公共卫生与预防医学系226019李文超汪徐林
南通大学公共卫生学院公共卫生与预防医学系(226019) 周 洁 张 晟 何 书 李文超 汪徐林 沈 毅
随机对照试验(randomized controlled trial,RCT)目前被认为是临床试验的金标准,但在实际工作中常受到伦理、经济等因素的影响,且因研究对象有严格的纳入排除标准,使其结论外推受到限制[1]。大样本观察性研究可部分弥补RCT的不足,但其研究过程未采用随机对照,基线以及重要预后因素在组间分布的不均衡可能会对结果的真实性造成影响[2]。倾向性评分匹配(propensity score matching,PSM)是一种均衡混杂因素的方法,近年来广泛用于大样本、非随机观察性研究[3-5]。目前用于实现PSM的软件为数不少[6-8],R语言的Matchit程序包以及Stata软件的psmatch2程序包就是专门用于实现PSM的易学易用的程序包[9-10]。本文拟通过实例展示如何在R与Stata软件上逐步实现PSM。
倾向性评分的概念与基本原理
倾向性评分(propensity score,PS)的概念是Rosenbaum 和Rubin 于1983 年首次提出,它是指将多个协变量的影响通过PS值的变化来表示[11]。根据PS值在组间进行分层、匹配、回归(协变量调整法)或加权分析,即均衡组间协变量的分布,最后在协变量分布均衡的匹配组内或层中进行效应值的分析[12]。
PS的基本原理是指在一定可观察协变量 (Xi) 的条件下,研究对象i(i= 1,2,…,n) 被分配到特定处理组(Zi= 1) 或对照组(Zi= 0) 的条件概率。此时,第i个研究对象被分配到处理组的概率可以表示为:
e(xi)=P(Zi=1|Xi=xi)
若给定的特征变量(xi) 与分组变量 (Zi) 是相互独立的,则:
e(xi) 即为PS值。
Rosenbaum 和Rubin 提出构建PS 模型需服从“强烈可忽略”(strongly ignorable)的假设,即计算PS值时,协变量应包含所有影响分组的混杂因素,或者说是不存在未识别的混杂因素[11]。它的使用具有两个条件:(1)条件独立性,即观察对象的分组选择只受纳入模型的协变量影响,而不受其他协变量的影响;(2)组间评分分布具有足够大的重叠区域,即样本量足够大且协变量的PS值相近。
在R软件上实现倾向性评分匹配
1.软件安装与程序包加载
R是一个自由、免费、源代码开放的软件,可用于统计计算、统计编程以及统计绘图[13]。截止至2017年7月25日,R软件最新版本为R3.4.1,用户可从官方网站http://www.r-project.org上获取最新的版本及相应的安装包。
R软件安装完毕后,还需要进一步安装和加载所需的Matchit程序包,具体安装与加载代码如下:
install.packages("Matchit")
library(MatchIt)
2.数据背景与加载
数据选择某医院2012年1月1日至2015年10月31日期间确诊的603例妊娠合并乙肝孕妇,探讨HBeAg对妊娠合并乙肝孕妇围产期不良结局的影响,其中暴露组(HBeAg阳性)172例,对照组(HBeAg阴性)431例。由表1可见,暴露组与对照组的基线资料无可比性,均衡性较差。
将数据以“HBV”为名、以“.csv”为格式存储在D盘里,使用“read.csv()”命令来读取数据,具体读取命令为:
HBV<-read.csv("D:/HBV.csv",header=TRUE,sep=",")
上述命令执行后,在命令栏中输入命令“HBV”,即可见到导入数据,表示数据加载成功。
3.倾向性评分匹配与结果输出
以孕妇是否为HBeAg阳性为分组因素,将其余变量作为协变量纳入PSM模型之中,并将结果存储在“m.out”这一新变量之中。匹配的算法选用了最常用的最近邻匹配算法(nearest neighbor matching),匹配的比例可通过设定ratio的数值来实现,一般设置为1~5,本例设置为1:1匹配。程序命令如下:
m.out=matchit(HBeAg~age+alt+ast+tbil+glu+wbc+cr+pt,data=HBV,method="nearest",ratio=1)
值得注意的是 PSM 中匹配算法的选择对结果的影响十分重要,本文列举了R中主要的匹配方法及其适用条件:
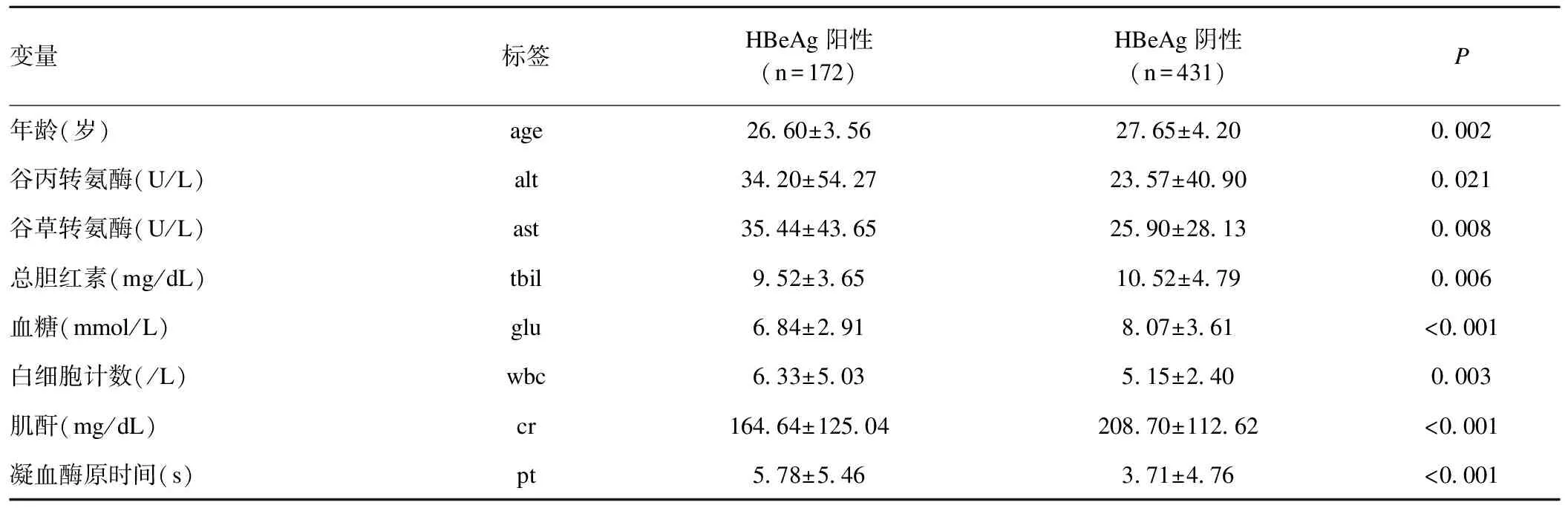
表1 孕妇基线特征表
精确匹配(exact matching)——该算法会精准地匹配每个协变量值完全相同的处理组与对照组。但当协变量较多或者协变量取值范围较大时,精确匹配可能较难实现。(method =“exact”)
最近邻匹配(nearest neighbor matching)——该算法将处理组与对照组中倾向性评分最接近的研究对象进行匹配。其优点在于可最大化匹配效果,最大程度上保留了研究样本的信息。(method =“nearest”)
优化匹配(optimal matching)——该算法的核心是尽量减少在所有配对组之间的平均绝对距离。需要注意的是使用优化匹配时,需要同时安装optmatch程序包。(method = “optimal”)
遗传匹配(genetic matching)——该方法是使用遗传搜索算法找出每个协变量的权重,使得匹配之后的样本达到全局最优平衡。缺点是当变量较多时,需要运行较长的时间。(method =“genetic”)
粗化精确匹配(coarsened exact matching)——这种算法可对特定协变量进行匹配,同时保持其他协变量的平衡。其优点是计算速度快,对多分类变量、极度不平衡资料以及多重填补后的缺失资料有着良好的适应性。使用粗化精确匹配需安装cem程序包。(method = “cem”)
接着我们可以对结果进行描述与绘图,命令如下:
summary(m.out)
plot(m.out,type="jitter")
plot(m.out,type="hist")
plot(m.out,type="QQ")
图1给出了匹配前PS模型基本信息(A)、匹配后PS模型基本信息(B)、均衡性检验(C)以及样本匹配结果(D)。首先匹配的基本信息包括了暴露组与对照组变量与距离(distance)的均值(means treated与means control)、对照组标准差(SD control)、暴露组与对照组的均值差(mean diff)以及QQ图中暴露组与对照组的中位数距离(eQQ med)、均数距离(eQQ mean)和最长距离(eQQ max)。接着均衡性检验提示,本例样本整体距离的均值差由原来的0.0713下降为0.0099,均衡性提升86.11%,所有变量均衡性提高50%以上,匹配效果良好。最后匹配结果包括了暴露组(treated)与对照组(control)匹配前(all)、匹配成功(matched)、匹配失败(unmatched)以及被排除(discarded)的样本量(sample sizes)。
图2是PS分布抖点图(jitter plot),它表示暴露组与对照组间匹配与未匹配者(unmatched treatment units:未匹配暴露组,matched treatment units:已匹配暴露组,matched control units:已匹配对照组,unmatched control units:未匹配对照组)PS的分布,从而了解匹配的效果。点的位置表示个体的得分情况;在1:n的情况下,点的大小则表示个体的权重。本例结果显示,获得匹配的个体倾向性评分较为接近,匹配效果较好。
图3是PS分布的直方图,表示暴露组与对照组间匹配前后(raw treated、raw control、matched treated、matched control分别指匹配前暴露组与对照组以及匹配后暴露组与对照组)PS值的分布,通过此图可看出暴露组与对照组匹配均衡性及匹配效果。本例中,匹配后对照组的PS由匹配前的不均衡变为均衡且与试验组接近,说明匹配效果较好。
图4为PS模型变量的QQ图,它表示暴露组与对照组间各个变量的PS分布,可看出单个变量的匹配前后的均衡情况。如年龄变量(age)的PS由匹配前的非正态分布变为匹配后的正态分布,说明年龄变量匹配效果较好。
4.完成匹配数据的导出
匹配完成后,需将成功匹配的数据导出进行效应值的分析。此时可使用match.data命令将数据存储在“m.data”这一新变量中,并使用“write.csv”命令将数据以“mHBV.csv”格式导出到D盘中,以便进一步的分析,具体命令如下:
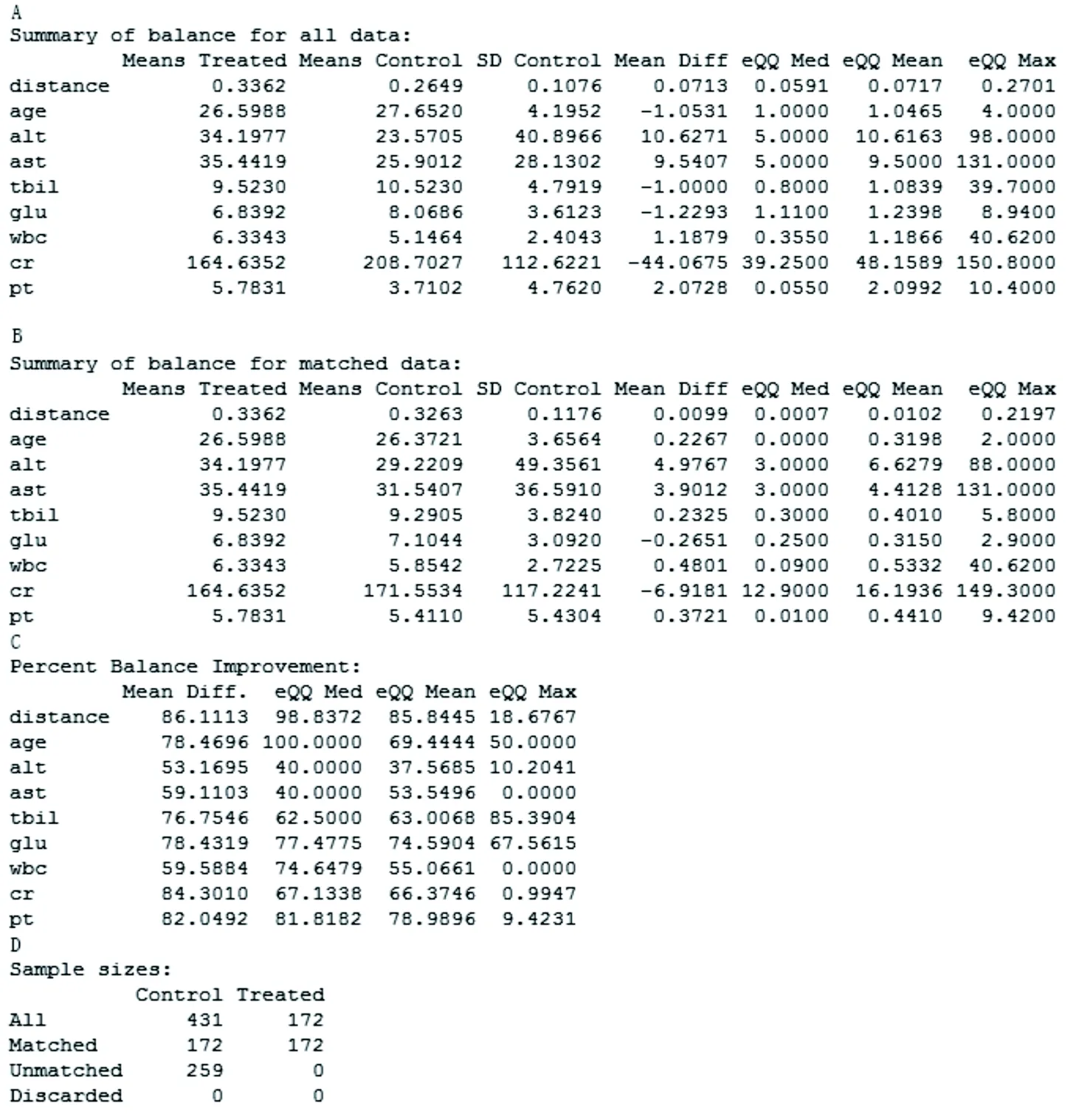
图1 倾向性评分模型结果、均衡性检验与样本匹配结果
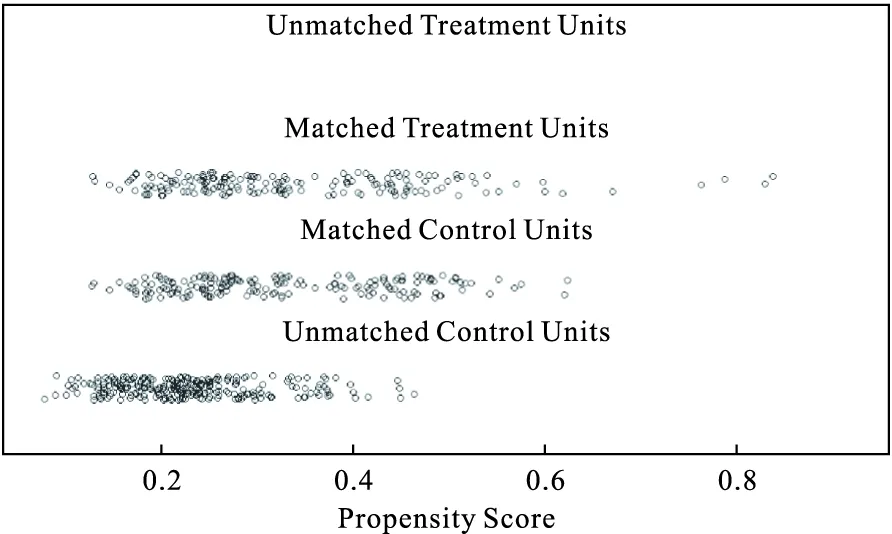
图2 倾向性评分分布抖点图
m.data<-match.data(m.out)
write.csv(m.data,file="D:/mHBV.csv")
数据导出后,软件自动生成了三个新变量:id为每一个观测对象唯一的ID号;distance为最近邻匹配的距离值;weight为匹配的权重值。
在Stata软件上实现倾向性评分匹配
1.Stata软件的下载与模块加载
Stata 软件是一套可进行数据分析、数据管理以及绘制专业图表的统计软件。用户既可通过窗口菜单进行操作,也可直接输入编程代码完成数据运算。用户可从 http://www.stata.com/stata14/ 购买最新版Stata14.0,本文使用的为该版本。
在 Stata 的命令栏输入ssc install psmatch2,即可自动搜索及安装psmatch2程序包。
2.数据录入
分析数据同前,可使用菜单栏中import过程来导入数据或者直接将excel 表格中的数据复制粘贴至数据编辑器之中。
3.倾向性评分匹配与结果输出
同样以孕妇是否为HBeAg阳性为分组因素,将其余变量作为协变量纳入PS模型之中。使用psmatch2命令,匹配的算法选用卡钳匹配算法(caliper),它是在最近邻匹配的基础上加一个限制条件,即暴露组与对照组PS差值需要在一定范围内才能进行匹配,卡钳值根据经验设为0.03[14]。匹配的比例可通过设定最紧邻算法后括号中的数值来实现,本例设置为1:1匹配。common选项可查看暴露组样本是否支持共同支撑假设,即强制排除暴露组中PS值大于对照组最大PS值或小于对照组最小PS值的样本。ties强制当试验组观测有不止一个最优匹配时同时记录。程序命令如下:
psmatch2 hbeag age alt ast tbil glu wbc cr pt,logit neighbor(1) caliper(.03) common ties
pstest,both
psgraph
Stata中的匹配方法主要为:最近邻匹配、核匹配(kernel matching)、局部线性匹配法(local linear matching)以及基于马氏距离的匹配方法(Mahalanobis matching)。[15]
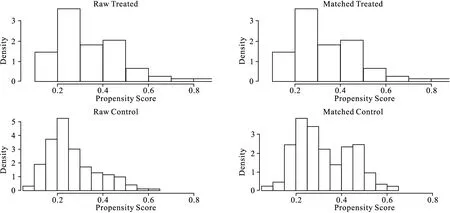
图3 倾向性评分分布直方图
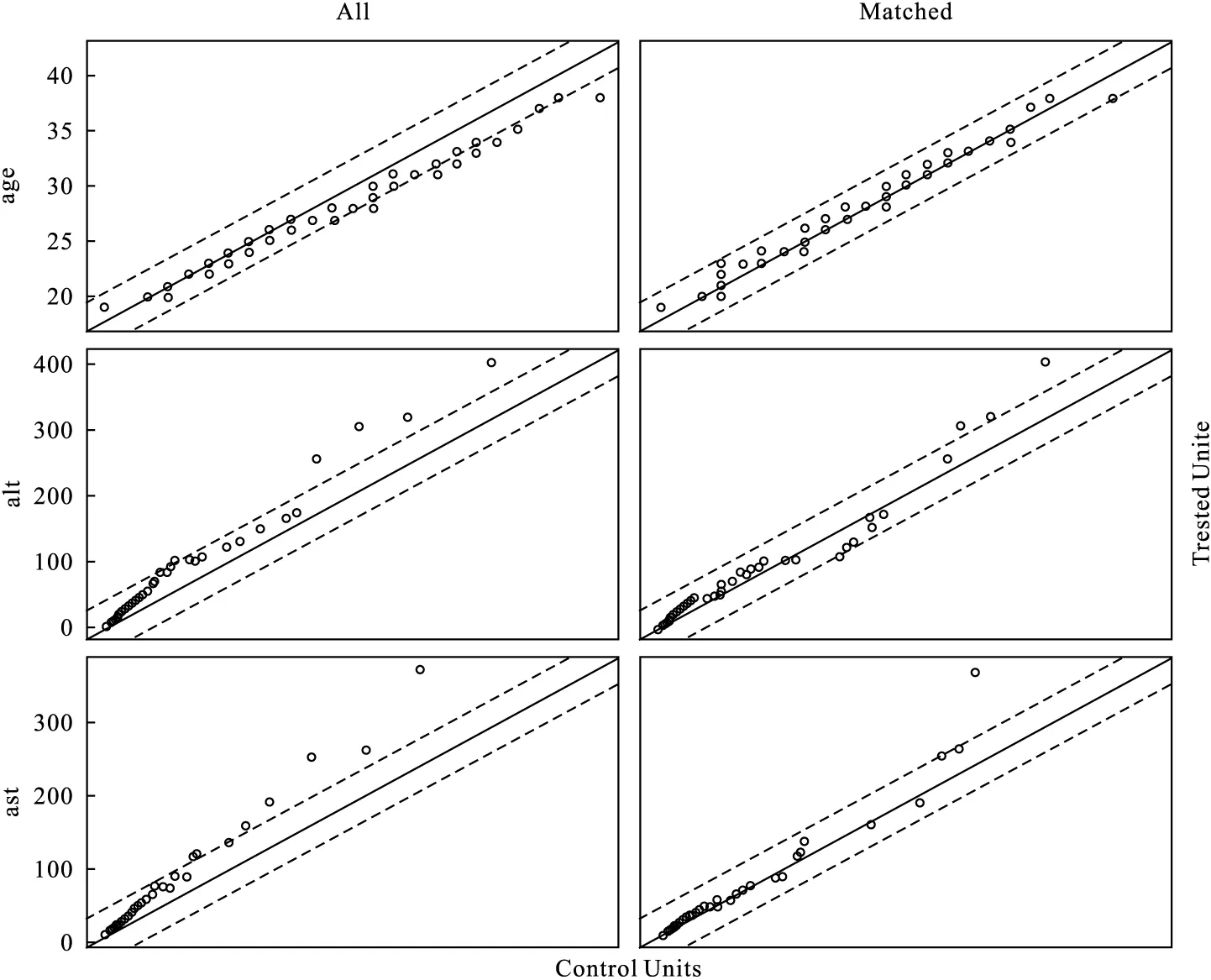
图4 倾向性评分变量QQ图*:只列举前三个变量(年龄,谷丙转氨酶、谷草转氨酶)
pstest命令可做出匹配后均衡性检验,理论上pstest只能对连续变量做均衡性检验,对分类变量应重新整理数据后运用χ2检验或者秩和检验。psgraph命令可对匹配结果进行图示。
首先psmatch2命令可以看到模型回归结果以及各变量的整体情况。接着打开数据编辑窗口,会发现软件自动生成了一些新变量,其中_pscore是观察对象的PS值;_id是自动生成的每一个观察对象唯一的ID号;_treated表示观察对象是否为暴露组;_n1表示的是观察对象被匹配到的对照的_id;_pdif表示已经完成匹配的观察对象间概率值之差。
pstest命令使用both选项可得到匹配前后的信息,均衡性检验结果可知,所有变量匹配后均由组间不均衡变为均衡,匹配效果良好(表2)。
psgraph命令作图结果可见匹配后样本倾向性评分值大致相同,匹配效果良好。与R不同的是,psgraph命令可绘制出变量是否具有共同支撑区域,本例中有部分暴露组样本被排除(图5)。
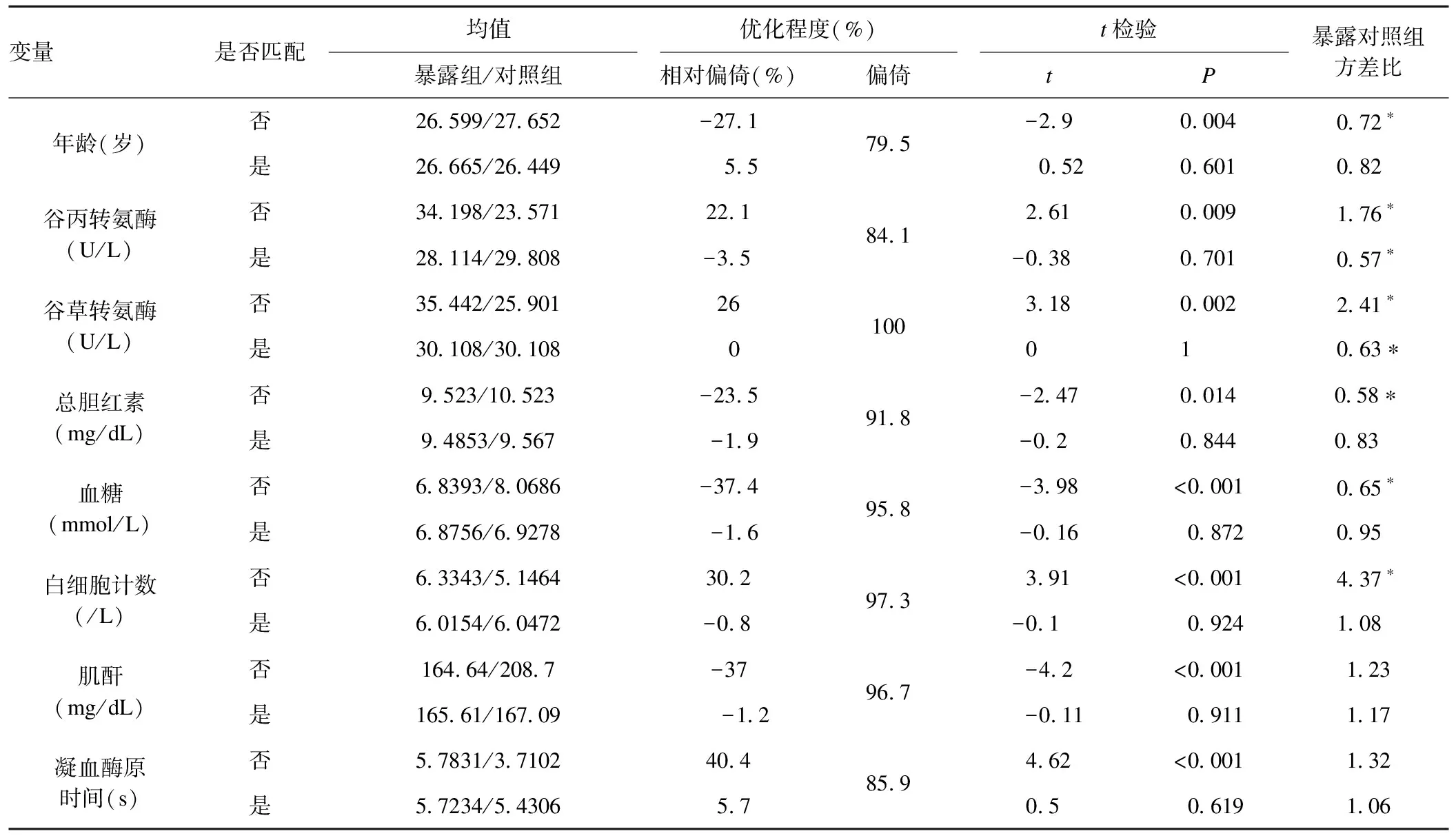
表2 倾向性评分模型均衡性检验结果
* 未匹配组方差比在[0.74,1.35]之外,匹配组方差比在[0.74,1.36] 之外。
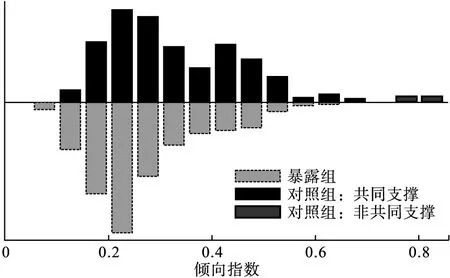
图5 倾向性评分模型匹配结果直方图
讨 论
PSM作为一种半参数统计方法,可以更加灵活地处理各种函数与模型,增加了暴露组与对照组合理匹配的可能性,限制也相对较少。尤其在处理较多混杂因素问题时,PSM与传统方法相比,运算更为便捷,匹配更为合理,可为研究者提供更高效客观的匹配选择。当然,PSM的应用也有其自身的限制。由于需要组间评分分布具有足够大的重叠区域,需要研究有一定的样本量支持,当样本量较小时,则无法解决协变量失衡的问题。另外,当数据存在缺失值,或有未知混杂因素时,此方法也难以使用。
目前,PSM的应用已经成为一个热点,但却缺少易学易懂易用的软件。SPSS软件在22.0之后的版本直接纳入了“倾向得分匹配”模块,虽然实现起来比较方便,但存在只能实现1:1匹配,匹配方法单一,没有绘图选项等缺点。如果研究者需要解决这些问题,则需要在SPSS中安装PS Matching模块,此模块的本质为调用R的Matchit包实现PSM,但此模块安装步骤较为复杂[7]。SAS软件同样在最新的9.4版本中STAT 14.3模块下加入了proc psmatch命令,使得实现PSM更加便捷。而之前的版本只能通过宏程序的方式来实现PSM[16]。
R的Matchit程序包与Stata的psmatch2程序包都有着简明的程序,直观的结果以及理想的绘图。R与Stata相比,有着更加多样的匹配选项和绘图选择;而Stata则可在匹配中选择共同支持区域,使结果更加合理。另外,它的均衡性检验可以直接比较匹配前后P值的结果,更直观方便。研究者可根据自身的需求选择来应用。
本文通过实例介绍了如何在R与Stata软件中实现倾向性评分匹配,并提供了详细的程序说明和结果解释,相信可以对运用PSM的科研工作者有所帮助。
