Heterogeneous Catalysis of CO2 Hydrogenation to C2+ Products
2018-09-18GAOYunnanLIUShizhenZHAOZhenqingTAOHengcongSUNZhenyu
GAO Yunnan, LIU Shizhen, ZHAO Zhenqing, TAO Hengcong, SUN Zhenyu
State Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, P. R. China.
Abstract: The increasing anthropogenic emission of CO2 leads to global warming, to address which three strategies can be considered: (1) decrease fossil fuel consumption through increased utilization efficiency and lower per capita consumption;(2) replace fossil fuels with renewable energy sources like wind, tidal, solar, and biomass energies; (3) utilize CO2 efficiently. Despite efforts to reduce energy use and increase the use of carbon-neutral biofuels, it seems that fossil fuels will continue to be a major energy source for the next few decades. Tremendous effort is therefore being focused on developing effective technologies for CO2 capture and transformation. In particular, the transformation of CO2 into fuels and chemicals via reduction with renewable hydrogen is a promising strategy for mitigating global warming and energy supply problems. The hydrogenation of CO2, especially to C2+ hydrocarbons and oxygenates, has sparked growing interest. The C2+ species can be used as entry platform chemicals for existing value chains, thus providing more advantages than C1 compounds. However, optimizing catalyst design by integrating multifunctionalities for both CO2 activation and C-C coupling remains an ongoing challenge. Here, we provide a timely review on the recent progress that has been made in the hydrogenation of CO2 to higher-order alkanes, olefins, and alcohols by various heterogeneous catalysts. The thermodynamics and kinetics, as well as possible reaction pathways for CO2 hydrogenation, are discussed. The hydrogenation of CO2 to hydrocarbons usually involves the initial generation of CO via a reverse water-gas shift (RWGS) reaction followed by hydrogenation of the CO intermediate. The RWGS reaction proceeds through a redox route and an associative pathway. “CHx” insertion (carbide-type) and “CO” insertion are two proposed mechanisms for this Fischer-Tropsch-like synthesis. Fe- or Co-based catalysts have been widely used to catalyze the hydrogenation of CO2 to C2+ hydrocarbons via the CO intermediate. C2+ hydrocarbons can also be obtained by combining CH3OH synthesis with the methanol-to-hydrocarbon process (MTH). This reaction pathway has been realized over bifunctional systems comprising a CH3OH synthesis catalyst and an MTH catalyst. Alternatively, CO2 hydrogenation can occur via a RWGS reaction to the CO intermediate, and subsequent formation of higher alcohols from syngas. Higher alcohols (mostly CH3CH2OH) have been produced by using a hybrid tandem catalyst. Understanding of the activation mechanism, precise C-C coupling, and synergy control between the two active components requires further research. In the final part, we describe the future challenges and opportunities in heterogeneous catalysis of CO2 hydrogenation. The combination of calculations (precise theoretical models) and experiments (in-situ spectroscopic techniques) will facilitate the design of advanced catalysts to achieve both high CO2 conversion and C2+ product selectivity.
Key Words: CO2 hydrogenation; Heterogeneous catalysis; C2+ species
1 Introduction
The incessant emission of carbon dioxide (CO2) into atmosphere resulting from combustion of fossil fuels gives rise to global warming1. This situation becomes more intense and unfavorable due to the increase in population, power plant,industries and energy consumption. The CO2level is estimated to increase up to 8 × 10−4(liter per liter air) during this century,and may reach 2 × 10−3(liter per liter air) by 2300, resulting in further temperature increase and ocean acidification2. Fossil fuels likely continue to be a main energy source up to 2050,and possibly beyond. The rapid consumption of fuel fossils also leads to upcoming depletion of natural carbon sources and global energy crisis3,4. From these scenarios, conversion of CO2 to valuable fuels and carbonaceous chemicals appears to be a promising strategy to ameliorate energy shortage and associated environmental problems5,6. The efficient utilization of CO2 as a feedstock of low (or even negative) cost can also create a new and sustainable “CO2economy”. However, CO2is inert and intrinsically stable (ΔGf0= −394.4 kJ·mol−1), thus requiring a substantial input of energy for transformation [~750 kJ·mol−1needed for dissociation of the C=O bond compared with that of C―C (336 kJ·mol−1), C―O (327 kJ·mol−1), and C―H (411 kJ·mol−1)].
Although reduction of CO2by using solar energy or electricity has attracted research interest, it is still long R&D necessary for practical implementation. Most of known electrocatalysts7–10or photocatalysts11,12only show activity for the formation of C1species, such as carbon monoxide (CO),methane (CH4), methanol (CH3OH), and formic acid(HCOOH). In addition, they usually have unsatisfactory efficiency and/or require the use of sacrificial reducing agents.Transformation of CO2by using high-energy hydrogen to yield hydrocarbons and/or oxygenates is one of the most intensively investigated areas13. Whether CO2conversion by hydrogenation contributes to a net reduction in CO2emissions is still a matter of debate, which seems to depend on the function of specific products and processes targeted.Nevertheless, CO2hydrogenation could be environmentally appealing to storing energy in chemicals and fuels with low carbon footprint when hydrogen is made from water electrolysis by using renewable or non-fossil resources.Homogeneous14and heterogeneous15catalysts have been applied to accelerate this slow kinetic process by reducing energy barrier. Homogeneous catalysts exhibit high activity and selectivity in CO2hydrogenation, but they suffer from drawbacks of high cost, and difficulty in recovery and regeneration. In contrast, heterogeneous catalysts are more chemically stable, cheaper, and easier to recycle, making them more promising for large-scale application in industry16,17.
In recent years, heterogeneous catalysis of CO2hydrogenation to C2+ hydrocarbons (up to C15 chain length)and/or oxygenates has garnered increasing research effort18,19,although this area still remains a grand challenge due to high C―C coupling barrier and the competition of C―C bond formation with the H―H and C―H bond formation. The C2+species can be used as entry platform chemicals for existing value chains, offering more advantages than C1 compounds.For example, C2–C4 alkanes increase the calorific value of natural gas or biogas through injection into gas-distribution grids. Higher (C5+) alkanes are liquids under ambient conditions, and have high energy densities and compatibility with existing liquid-fuel distribution and end-use infrastructures. Hence, they are attractive precursors of jet fuels. C2–C4 light olefins are valuable base chemicals for the production of polymers20. Relative to CH3OH, higher alcohols have been regarded as alternative precursors for short-chain olefins as well as better and cleaner fuels because of their high octane numbers and low emissions of NOx, ozone, CO, and volatile aromatic vapors after combustion21.
Many excellent reviews and perspectives on the conversion of CO2are available15,20,22–24. Nonetheless, most of the reviews focus on the production of C1products (e.g., CH4,CH3OH and its derivatives)25–27. Here we examine how CO2hydrogenation occurs to generate higher-value C2+chemicals, particularly long-chain alkanes, olefins and alcohols. Relationships between structure and activity in efficient heterogeneous catalyst design are discussed. A foundational summary for possible reaction pathways is provided. An outlook of future opportunities for conversion of CO2into C2+fuels via hydrogenation is also described.
2 Mechanistic understanding of CO2 hydrogenation to C2+ species
The reverse water-gas shift (RWGS) reaction [Eq. (1)] is key in CO2hydrogenation. This reaction enables transformation of refractory CO2into more reactive CO by introducing hydrogen.Such endothermic reaction (ΔH298K= 41.2 kJ·mol−1, ΔG298K=28.6 kJ·mol−1) is remarkably milder than the production of CO by breaking the C―O bond in CO2 which requires high temperatures (> 2000 °C).

The CO, produced by the RWGS reaction, could be further hydrogenated to hydrocarbons and alcohols [Eqs. (2)–(4)]28. It can also be converted back into CO2 and hydrogen in the presence of water following a water gas shift (WGS) reaction pathway. A direct route from CO2to C2+hydrocarbons has been also proposed, which however unlikely plays a major role29.Despite this being the case, the direct hydrogenation of CO2to CH4(Sabatier reaction) is possible [Eq. (5)], which consumes the largest use of hydrogen per CO2 (ΔH298 K = −252.9 kJ·mol−1, ΔG298 K = −130.8 kJ·mol−1).
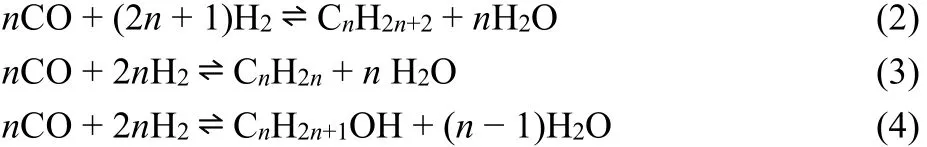
where n is the stoichiometric coefficient.

Compared with traditional Fischer-Tropsch (FT) synthesis,this two-step reaction requires nearly double amounts of hydrogen and generates more water, a byproduct which may deactivate catalyst and slow down the reaction. The conversion of CO2 by the RWGS reaction is limited due to relatively low temperatures employed in the second step of FT reaction. Only 13%–23% of CO2is converted into CO at temperatures between 220 and 300 °C with a H2/CO2ratio of 3(stoichiometric ratio for CO2hydrogenation to ―CH2―)30.The maximum conversion of CO2 ranges from 10% to 50% at reaction temperatures from 200 to 500 °C.
Calculations by using subroutine RGIBBS along with Soave-Redlich-Kwong model showed that above 550 K,increasing reaction temperature had a detrimental effect on the equilibrium conversion of CO2and H2. C2compounds were the major products within the temperature range 350–850 K in a C1-free system, while the formation of longer chain hydrocarbons was favored with the increase of temperature up to 750 K, in a similar way to increasing reaction pressure(within 1 and 30 bar) based on polymerization FT mechanism.CO2equilibrium conversion was promoted by increasing H2/CO2 ratio, reaching 100% when the ratio was > 4.231.However, the H2/CO2 ratio was shown to have a marginal effect on the yield of hydrocarbons. It was calculated that the addition or in situ removal of water in the tandem system did not affect the equilibrium conversion of CO2or the formation of hydrocarbons. The introduction of CO into a CO2/H2mixture keeping constant COx/H2 molar ratio (1 : 3) enhanced the hydrogen yield into hydrocarbons but had a small effect on CO2 equilibrium conversion at CO/CO2molar ratios less than 0.520.In the case of higher ratios, the CO2equilibrium conversion decreased despite increasing the overall COxequilibrium conversion due to the conversion of CO into CO2through WGS reaction. We should point out that these theoretical results on reaction conditions for the tandem process need to be further confirmed by experiment.
The formation of C2+hydrocarbons and alcohols, albeit being less likely than that of CH4, is more thermodynamically favorable than that of CH3OH at low temperatures (Fig. 1a).Zhang and co-workers performed a thermodynamic study, and investigated the ΔG for CO and different hydrocarbons as a function of reaction temperature (Fig. 1b). The variation of temperature was observed to have the minimum effect on the generation of CH4. At temperatures below about 300 °C (ΔG <0 for olefins), increasing temperature favored the yield of olefins in the equilibrium products. For the synthesis of C2–C4 olefins at CO2 conversion of 72.8%–74.5%, optimal reaction temperatures were proposed to be in the range 300–400 °C,pressures in the range 2.0–3.0 MPa, and H2/CO2feed gas molar ratio of 332.
3 Possible reaction pathways
3.1 Synthesis of alkanes and olefins
3.1.1 CO2 hydrogenation via CO intermediate
The hydrogenation of CO2to hydrocarbons usually involves the initial generation of CO via a RWGS reaction. Two important reaction mechanisms on the RWGS reaction have been put forth, namely a redox route and an associative pathway. In the redox mechanism, CO2is adsorbed and activated on reduced metals or metal oxides to form CO.Hydrogen acts as a reductant, and does not participate in the formation of intermediates in the RWGS reaction. This redox mechanism has been confirmed by XPS analysis of the oxidation states of Pt/TiO2catalyst33. The associative mechanism is related with the decomposition of intermediates such as surface formate, carbonate, or carboxyl species that are derived from the association of hydrogen with CO2. The type of intermediate depends on reaction conditions and the catalyst used34.
In the following FT reaction, formed CO reacts with unconverted H2. However, the pathway of chain growth in F-T synthesis remains controversial. “CHx” insertion (carbide type)35and “CO” insertion36are two proposed mechanisms. The former assumes CO activation either by direct reaction with chemisorbed hydrogen or H-assisted pathway possibly via formyl (HCO*) and formaldehyde (H2CO*) or hydroxymethylene (HCOH*) species to form surface CHx*monomer (Fig. 2). The direct CO activation removes O*from CO2, while the H-assisted CO dissociation forms H2O exclusively. The H-assisted route is likely a predominant kinetically-relevant step on Co catalysts as confirmed by experimental measurements and DFT calculations37.

where Wnrepresents the weight proportion of products with n carbon atoms in their chain, α stands for the chain-growth probability, and n is the number of carbon atoms. It has been predicted that the fraction of C5–C15 reaches a maximum of 60% at α = 0.8. Deviations from the ASF model were observed on Co-based catalysts. This is due to the possibility that CO2 conversion took place through a separate methanation pathway rather than via F-T synthesis. Nevertheless, the α parameter in liquid hydrocarbon products provides a suitable basis for comparison of various product distributions40.
3.1.2 CO2 hydrogenation via methanol intermediate
C2+hydrocarbons can also be obtained through an initial formation of CH3OH [Eq. (7), ΔH298K= −49.5 kJ·mol−1]followed by its subsequent transformation.

This reaction pathway has been recognized over bifunctional systems comprising a methanol synthesis catalyst and a methanol-to-hydrocarbon (MTH) catalyst. In a recent study41,CHxO species (mainly surface CH3O*, CHO*species, and gas-phase CH3OH) were found to be generated via CO2hydrogenation on ZnZrO. These derived CHxO species migrated into a Zn-modified SAPO-34 zeolite to convert into lower olefins (Fig. 3).
Two reaction mechanisms for CH3OH formation have been proposed including a formate route and a RWGS pathway. In the formate route, CO2is hydrogenated step by step to CH3OH directly. Whilst in the RWGS pathway, CO2is firstly reduced to CO at a metal-support interface, and then consecutivelyhydrogenated to CH3OH. CO are released in this process as well, that is why CO is always associated with CH3OH production.
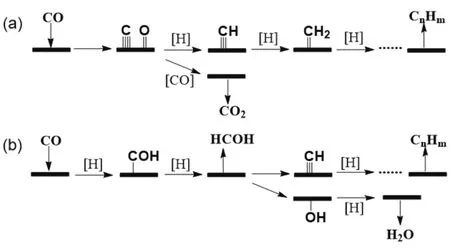
Fig. 2 Possible pathways for the formation of hydrocarbons from CO 37∶ (a) direct CO activation pathway on Fe catalysts, and (b)H-assisted pathway on Co catalysts.Adapted from Elsevier publisher.
Fig. 1 (a) Evolution of the standard Gibbs reaction free energy for the hydrogenation of CO2 into n-alkanes (○) and terminal n-alcohols(□) 20. (b) Effect of reaction temperature on Gibbs free energy of CO2 hydrogenation to hydrocarbons and oxygenates 32.Fig. (a) Reproduced with permission from © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
The MTH process over zeolite catalysts, was first discovered by Mobil Research Laboratories in 197642. Medium-pore zeolites were reported to produce C5–C11hydrocarbons,whereas small-pore molecular sieves generated C2–C4hydrocarbons. The distribution of hydrocarbon products is affected by the structure and acidity of zeolite. Possible mechanistic proposals involve oxonium ylide mechanism,carbine mechanism, carbocationic mechanism, free radical mechanism, hydrocarbon pool mechanism, etc.22.
3.2 Synthesis of high alcohols
CO2 hydrogenation can proceed through a RWGS reaction to CO intermediate, and subsequent the formation of high alcohols from syngas43,44. Na in association with the carbide form of cobalt was speculated to enhance the production of ethanol from CO through a CO insertion mechanism, as illustrated in Fig. 4a45. Whilst on iron catalysts, CO was hypothesized to dissociate, forming adsorbed oxygen (Oad),which reacted with adsorbed hydrogen, to produce chemisorbed OH (OHad). The OHadthen reacted with hydrocarbon species such as alkylidenes (R―CH2―CH=) to yield ethanol, as shown in Fig. 4b46.
Alternatively, on Rh10Se/TiO2catalyst, the evolution of ethanol was suggested to occur from direct CO2hydrogenation rather than CO hydrogenation47. Specifically, CHx,adsspecies on Rh reacted with COy,ads to form acetate, which was further hydrogenated to ethanol on Rh sites.
4 Catalysts of CO2 hydrogenation to C2+species
4.1 C2+ alkanes
The majority of heterogeneous catalysts for CO2hydrogenation to alkanes are Fe-, and Co-based catalysts, asshown in Table 148–71.
Fig. 3 Schematic of CO2 hydrogenation reaction mechanism on ZnZrO/SAPO 41.Reproduced with permission from © 2017 American Chemical Society.
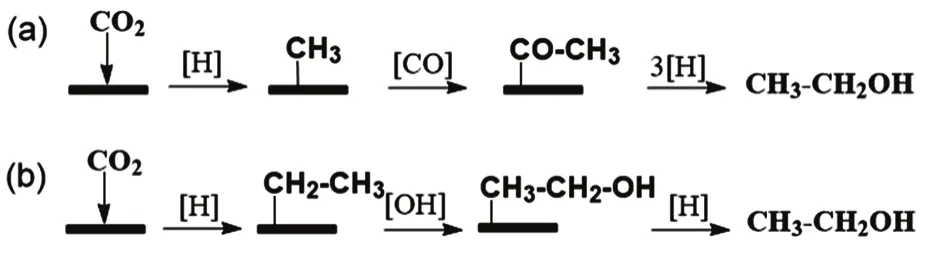
Fig. 4 Proposed pathways for the formation of ethanol over(a) Na-doped Co catalysts 45, and (b) Fe/NCNT catalysts 46.Adapted from Elsevier publisher.

Table 1 Reported catalytic systems for the synthesis of C2+ alkanes by CO2 hydrogenation.
4.1.1 Fe-based catalysts for both RWGS reaction and Fischer-Tropsch reaction
Fe-based systems can catalyze both RWGS reaction and the hydrogenation of CO to alkanes72,73. The Fe3O4and Fe2O3phases of Fe catalysts can be reduced to Fe5C2, which is considered to be the active phase for the formation of long-chain alkanes74.
4.1.1.1 Promoters
Bulk Fe catalyst in the absence of promoters presents some limitation in terms of stability, activity and selectivity to hydrocarbon products. Nevertheless, the addition of promoters such as alkali metals (Na, K, or Rb)75,76and transition metals(Mn, Cu, La, Zr, Cr, Mo, or Ta)30,55,77can remarkably enhance resistance to deactivation, and improve selectivity to long-chain alkanes. Alkali elements boost catalytic activity by acting as (i)an electronic promoter to shift product distributions to heavier hydrocarbons, (ii) a promotor of CO2 adsorption through the introduction of basic sites, and (iii) an inhibitor of surface hydrogenation. Tuning the content of promoters may increase the olef i n to paraff i n ratio and the average molecular weight of the products78. Among the alkali promoters, K was reported to be the most effective one79. Besides those functions listed above, K may also increase the content of Fe5C2, and shifted Fe in the oxide and carbide phases to a more reduced state, thus increasing the relative exposure of active sites on the catalyst surface80. Recently, Na modified Fe-Zn catalysts were synthesized through a microwave-assisted hydrothermal method by using a Na-containing ZnFe2O4precursor (Fig. 5a)56.Compared to bare Fe2O3, physically mixed ZnO-Fe2O3, and Na-free Fe-Zn catalysts, the ZnFe2O4-derived catalyst with a Na content of 0.08% (w) or beyond were superior, giving rise to a high CO2conversion, an improved C5+liquid-fuel selectivity (~58%), and a high olefin to paraffin ratio (~11) in CO2hydrogenation (Fig. 5b).
Transition metals such as Mn can promote the reduction of Fe catalysts, and increase the dispersion of catalyst particles as well as the catalyst surface basicity. Relative to Mn, Zn presents a more basic surface, and enables higher CO and CO2adsorption. Zn also plays a role in stabilizing a catalyst, thereby limiting its initial deactivation, and granting better yields to alkanes than Mn76. The incorporation of other transition metals including Co, Ni, Cu, as well as noble metals such as Pd in Fe catalysts also enhanced the yield of C2+alkanes from CO2hydrogenation81.
Metal oxides can also serve as structural and electronic promoters68. It was reported that SiO2, TiO2, ZrO2, HfO2, CeO2 and MnO2 might modify the texture and surface reactivity ofK/Fe-Al-O catalyst, and therefore improved their performance in CO2hydrogenation (Fig. 6). The SiO2additive existed in the form of Fe-carbide, and selectively blocked the Fe-sites. While ZrO2existed in the form of grafted species (residual Fe-Al-O spinel), and oxide nanoparticles (NPs), which contributed to improved activities for both the RWGS reaction and the FT synthesis. This was attributed to the strong electronic interactions between ZrO2and neighboring metallic Fe-carbide phase.
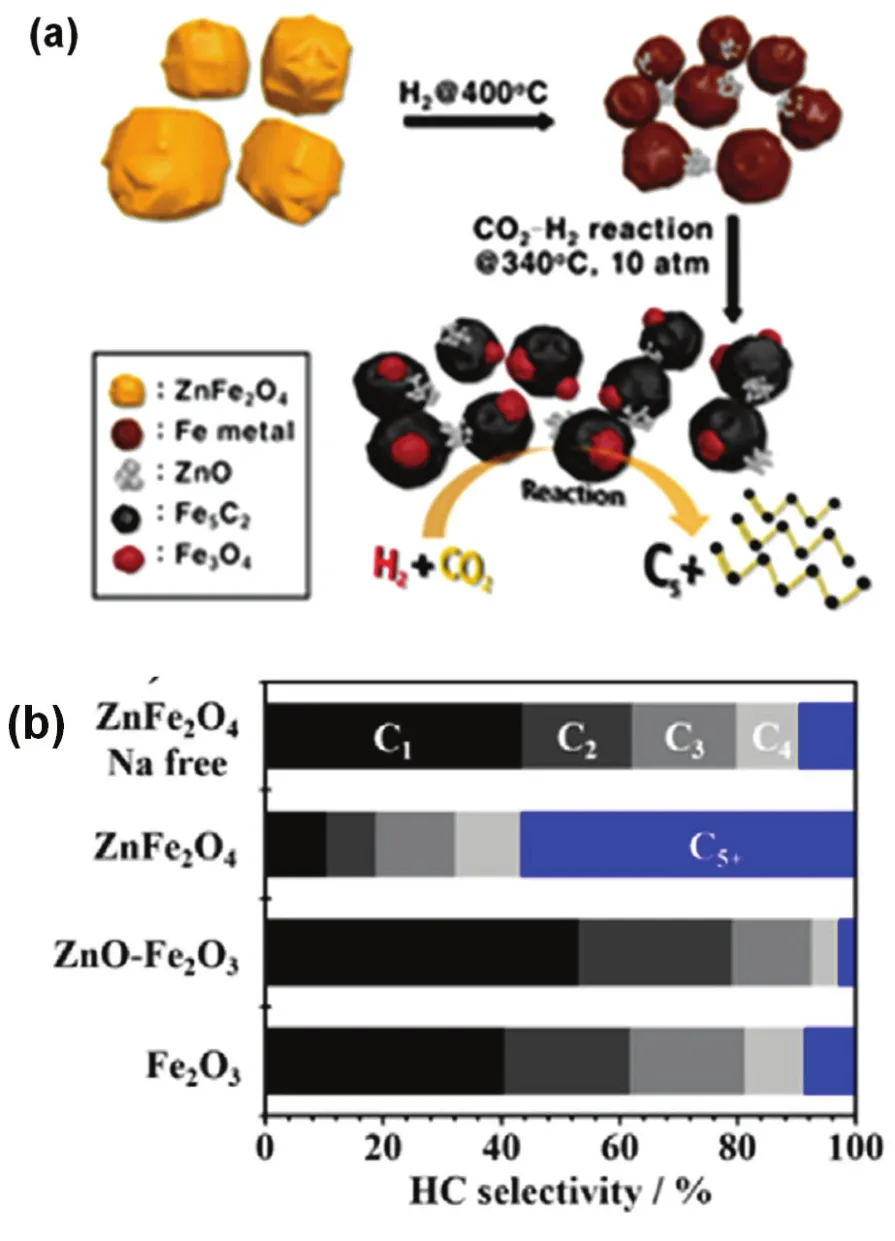
Fig. 5 (a) Structural evolution and mechanistic idea of ZnFe2O4-derived catalysts for CO2 hydrogenation to yield C5+ liquid fuels.(b) CO-free hydrocarbon selectivity on Fe2O3, ZnO–Fe2O3, ZnFe2O4, and Na-free ZnFe2O4 catalysts 56.Reproduced with permission from © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
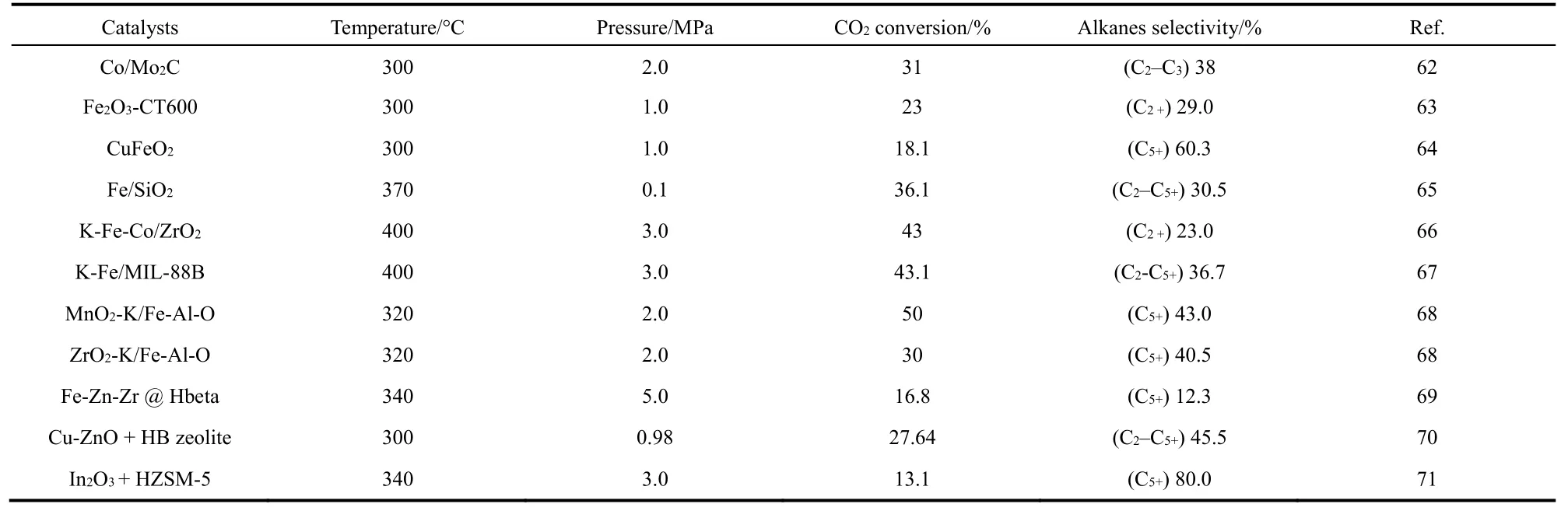
continued Table 1
4.1.1.2 Supports
Supports can offer high surface area, help to disperse the active components, and make the catalyst mechanically strong enough for long-time operation. A proper interaction between supports and the active sites can influence the whole process to a large extent82.
Metal oxides, especially SiO2, Al2O3, TiO2, CeO2and ZrO2,are the most commonly used supports for Fe-based catalysts83,84,54. Al2O3 exhibits abundant surface Al-OH groups, which favor the adsorption of CO intermediate. The large surface area of Al2O3facilitated the formation of small iron carbide particles with high dispersion, affording C5+hydrocarbons with selectivity of up to 31.1%50. It was found that only an appropriate pore size of Al2O3falling in the range 7–10 nm could lead to optimum activities57. Oxygen defects on reduced and monoclinic ZrO2 (m-ZrO2) outperformed better than tetragonal ZrO2(t-ZrO2) in adsorption and activation of CO285. Murciano et al.58investigated the effect of the morphology of ceria supports on the catalytic properties of Fe-based catalysts. Among the three kinds of morphologies(particle, rod and cube), nanostructured ceria rods provided the highest hydrocarbon selectivity, while ceria cubes presented the highest olefin/paraffin ratio.
Some other supports, like carbon nanotubes (CNTs), Mo2C,zeolite, MCM-41, and metal-organic frameworks (MOFs) have also been used in Fe catalysts69,86. CNTs have high specific surface area, multitudinous pore structure, superior chemical inertness, and eminent recycling characteristics. Fe NPs deposited on CNTs were reported to display low deactivation rates87. Surface modif i cation of CNTs with nitrogen or oxygen is usually employed to provide strong anchoring sites for catalyst particles. Iron oxide NPs supported on N-doped CNTs were observed to be reduced more easily than those on O-doped CNTs51,52. Another interesting support is MOFs which have advantages such as large accessible surface areas,high porosity, and tunable functionalities. MOFs composed of abundant organic struts and regularly arranged metal nodes, can be transformed to carbon materials decorated with nano-structured metal species after annealing. As an example,porous carbon supported Fe NPs were synthesized via one-step pyrolysis of Fe-MIL-88B67. This catalyst displayed good selectivity to C5+products (19.2%), and stability for CO2hydrogenation.
4.1.2 Co-based Fischer-Tropsch catalysts
Co catalysts are widely used in F-T synthesis particularly for the production of heavier hydrocarbons. It was suggested that Mn-modified Co2C was the active phase to the formation of long-chain hydrocarbons88. While Co has very low RWGS reaction activity, and acts primarily as a methanation catalyst when CO2 is used instead of CO89. To suppress methanation and improve catalytic performance of Co catalysts for CO2 hydrogenation, the selection of promoters and supports is of importance.
4.1.2.1 Promoters
Note that unmodified cobalt systems tend to generate > 90%CH4 in CO2 hydrogenation90,91. To reduce the production of CH4, small amounts of promoters including Li, Na, K, Mo, Cr and Mn were often added to Co-based catalysts45,53,92. For example, upon introduction of K to CoCu/TiO2catalyst, CH4formation was suppressed, and the main product was C5+. But CO2conversion decreased, and CO selectivity increased59.Similarly, the addition of Mo and Na to Co enhanced the selectivity of C2+ alkanes60.
4.1.2.2 Supports
Similar to Fe-based catalysts, the use of support (TiO2, CeO2,SiO2, MgO and ZSM-5) affects the size of Co crystallites and their activity by providing a high surface area and metal-support interactions. For Co-Na-Mo catalysts, C5+hydrocarbon selectivity was in the order of ZrO2 < TiO2 <Al2O3< CeO260.
4.1.3 Bifunctional catalysts for both methanol synthesis and methanol-to-alkane transformation
Alkane products can also be obtained through cascadecatalysis combining methanol synthesis from CO2hydrogenation, and a subsequent MTH reaction70,93. Typical catalysts for the methanol synthesis from CO2/H2 include Cu-based materials (Cu supported on ZnO94, Ga2O395, ZrO296)and noble metals (Pd97, Pt98, Au99, Ag100), while the commonly used catalysts for the MTH process are zeolites101.

Fig. 6 Effects of additives on the catalytic performance of K/Fe-Al-O spinel in CO2 hydrogenation∶ (a) selectivity to C2–C4 hydrocarbons, and (b) selectivity to C5+ hydrocarbons 68.Reproduced with permission from © 2017 Royal Society of Chemistry.
A number of highly efficient bifunctional catalysts have been reported. Wang et al.69developed a Fe-Zn-Zr@zeolite core-shell catalyst, with Fe-Zn-Zr as the core and the zeolite(HZSM-5, Hbeta, and HY) as the shell. The Fe-Zn-Zr core promoted methanol synthesis, and also suppressed the unwanted RWGS reaction. While the zeolite shell provided a confined reaction space, and acidic sites to convert methanol into isoalkanes. The catalyst with a double-zeolite (HZSM-5 and Hbeta) shell exhibited even higher performance for isoalkane synthesis, resulting from a synergistic effect between the two zeolites. A bifunctional catalyst comprising In2O3and HZSM-5 was reported, which enabled selective production of liquid fuels with C5+fractions as high as 78.6% from CO2hydrogenation (Fig. 7)71. The oxygen vacancies on the In2O3surface benef i ted CO2 activation and hydrogenation. The undesirable RWGS reaction was inhibited by moving the two components into a closer proximity (from Fig. 7b to Fig. 7d),yielding a high selectivity for hydrocarbons.
4.2 C2+ olefins
Synthesis of lower olef i ns from CO2hydrogenation has achieved advances in recent years102,103. Relevant solid catalysts are summarized in Table 241,103–116. The formation of short-chain olefins is always accompanied with CH4and >C4hydrocarbons117. Despite that a high selectivity of C2–C4olefins up to 65.2% has been obtained at low CO2conversion(25.7%), further improvement is still needed to control the chain growth.
4.2.1 Catalysts for both RWGS reaction and CO hydrogenation
As the case of alkane production, light olefins can be produced from CO2through a combined route of RWGS reaction and subsequent CO hydrogenation. A catalyst needs to be active for both RWGS reaction and CO hydrogenation under the same operating conditions. Fe-based catalysts are predominantly used for the synthesis of C2–C4olefins104,63,118.
4.2.1.1 Promoters
Fe catalysts alone show insufficient selectivities to olefins.This can be addressed by adding promoters119. The promoterscan enhance structural and chemical properties, and stabilize active components120. In addition, these promoters were supposed to increase CO adsorption while decrease hydrogen adsorption on the catalytic surface, thereby increasing the surface coverage of dissociatively adsorbed CO. A high CO coverage can inhibit the olefin readsorption rate, leading to higher olefin selectivity105. Alternatively, a strong interaction between an active phase (such as Fe2O3) and a support (such as K2O), may favorably suppress the hydrogenation of olefins113.

Fig. 7 Inf l uence of the integration manner of the active components on catalytic behaviours under the same conditions. (a)Dual-bed conf i guration with In2O3 packed below HZSM-5 and separated by a layer of quartz sand. (b) HZSM-5 packed below In2O3 and separated by quartz sand. (c) Stacking of granules with the In2O3,HZSM-5 and quartz sand particle sizes of 250–380 µm. (d) In2O3 and HZSM-5 particles well mixed without quartz sand. (e) In2O3 and HZSM-5 mixed with an agate mortar 71.Reproduced with permission from © 2017 Nature Publishing Group.

Table 2 Reported catalytic systems for the synthesis of light olefins (C2–C4) by CO2 hydrogenation.
The addition of alkali metals (Li, Na, K, Rb and Cs) to Fe-based catalysts could improve the selectivity of C2–C4olefins. The yield of C2–C4 olefins increased in the following order: Fe < Li+-modified Fe < Na+-modified Fe < Cs+-modified Fe < K+-modified Fe < Rb+-modified Fe121,103. Cheng et al.122investigated the effect of Na on the structure and catalytic performance of supported Fe catalysts. Olefin selectivity increased with the amount of Na, while alkane selectivity decreased inversely. It seems that Na can promote the surface basicity of catalyst, which favors for olefin production.
In addition to alkali metals, group 11 metals (Cu, Ag, and Au) and group 13 metals (Al, Ga, and In) can also act as promoters106. Among the Group 11 metals tested, Au in Fe/SiO2catalyst performed the highest selectivity of C2–C4.However, In was far more effective than the others, affording a C2–C4 olefin selectivity of 65.2% upon the addition of 3%(mass fraction) In to the system. Theoretical investigations suggested that In2O3promoted CO2activation and hydrogen adsorption through a dissociative process123. Meanwhile, In2O3was also active for the dehydrogenation of propane by utilising CO2 as a mild oxidant, and high levels of C2–C4 olefins were attained124.
Ce, La and Mn have also been used to modify Fe catalysts107.Mn may function as both structural and electronic promoters,imparting improved activity and selectivity toward light olef i ns55,125,126. The addition of CeO2in Fe/Mn/K catalyst resulted in an approximately 22% increase in CO2 conversion,and a 5% increase in olef i n formation127. It was speculated that CeO2accommodated large amounts of hydrogen within its lattice. Smaller CeO2particles had more lattice defects/oxygen vacancies, thus being more easily reduced than those larger ones. Tuning the particle size of CeO2allowed one to tailor the catalyst’s activity and selectivity.
Other reported metals to promote CO2 conversion include Cr and Zn128,129. Zn likely increased catalyst surface basicity,enhancing selectivity for C2–C4olefins130.
4.2.1.2 Supports
The widely used catalyst supports include SiO2, Al2O3, TiO2,ZrO2, mesoporous carbon, and CNTs. Among these supports,ZrO2 supported K-Fe catalyst provided the highest selectivity(42.3%) and yield (13%) to lower olef i ns. Two aspects may be relevant. On the one hand, ZrO2offered adsorption sites for surface reaction intermediates. On the other hand, the synergistic effect of K and ZrO2hindered the re-adsorption of olef i ns, improving CO2 hydrogenation activity towards light olefins103. Graphitic carbon can enhance electron abundance on those metal-rich sites, resulting in high activity. The inner concave surface of graphitic carbon both Fe3O4@carbon core-shell nanostructures and Fe NPs encapsulated inside the lumen of CNTs, was calculated to be able to improve the reducibility of iron oxides relative to the outer convex surface131. Graphitic carbon shells exhibit lower energy barriers for the surface migration of hydrogen as compared with other supports like Al2O3or SiO2, which in turn leads to lower selectivity of olefin51. Fe, Fe5C2and Fe3O4encased in partially graphitized carbon shells were synthesized by Gupta and co-workers and examined for CO2 hydrogenation (Fig. 8a).The olefin/paraffin ratio (O/P) was found to increase when the carbon content was increased from 0.5 to 1.5. However, further increasing the carbon content by deposition of excess carbon around the Fe3O4core probably hindered the accessibility of reactants to the Fe active sites, and reduced the O/P (Fig. 8b)108.
ZSM-5 zeolite with suitable acidity and pore size can also act as a support for Fe catalysts to improve the selectivity of light olefins109. MOFs, such as ZIF-8 can be stable up 300 °C,making such porous materials a potential support for high temperature and high pressure catalytic reactions. ZIF-8 compares more favorably than MIL-53(Al) and γ-Al2O3 supports in terms of CO2 conversion and olefin selectivity110.Larger ZIF-8 crystals corresponded to lower olefin selectivitydue to occurrence of secondary reaction of hydrogenation during the internal diffusion process.

Fig. 8 (a) A cartoon of Fe3O4@carbon core-shell nanostructure.(b) Fe time yield (FTY) and O/P ratio of catalysts with different carbon contents in CO2 hydrogenation 108.Reproduced with permission from © 2016 WILEY-VCH Verlag GmbH & Co.KGaA, Weinheim.
4.2.2 Bifunctional catalysts for both methanol synthesis and methanol-to-olefin transformation
Hybrid catalysts that are active for both methanol synthesis and MTH conversion have been also used for the production of olefins. Zeolites with weak acidic sites and narrow micropores may facilitate the selective formation of light olefins. SAPO is such kind of zeolite132, which could yield C2–C4 olefins with selectivities of up to 80%111. Indeed, a ZnZrO/SAPO catalyst(Fig. 9a,b) gave 80% selectivity of C2–C4olefins, 14% C2–C4alkanes, 3% CH4, and 3% C5+among all hydrocarbon products at a CO2conversion of 12.6% (Fig. 9c)41. CO2conversion increased with increasing reaction temperatures, but the olefin selectivity decreased. This is due to the fact that higher temperatures favor RWGS reaction. To attain a high yield of lower olefins, the reaction conditions were optimized with reaction temperature of 380 °C, space velocity of 3600 mL·g−1·h−1.
4.3 High alcohols
High alcohols (C2+OH), relative to CH3OH are more desirable products as neat fuels, fuel additives, and hydrogen resources in fuel cells133,134. Recent years have seen great advances in the synthesis of high alcohols with CO2by homogeneous catalysis135−138. In the following part, we focus on significant achievements that have been made on heterogeneous catalysis of CO2hydrogenation towards high alcohols. Recent advances in this regard are summarized in Table 343,76,139-148.
Noble metals (Rh, Pt, Ru) and transition metals (Cu, Fe, Co)on metal oxide supports (e.g., Fe2O3, Co3O4, ZrO2, Al2O3,SiO2) have been widely used for CO2hydrogenation to synthesize high alcohols134. As opposed to single metals,bimetallic catalysts (such as CuFe, CuZn) can create a synergism between the two metals, leading to higher catalytic efficiency139,140,76. A MoCoK sulf i de catalyst showed a potential for C2+alcohol production with C2+alcohol selectivity of 10.9% from CO2hydrogenation. In this hybrid catalyst, Coserved as an electron-donating additive, and shifted the binding energy of Mo 3d to a lower value. Such shift was related to the interaction between Mo and Co. It was found that the interaction between Co and Mo was enhanced when using activated carbon as a support, whereas the interaction became weakened in the case of SiO2, Al2O3, and TiO2141.
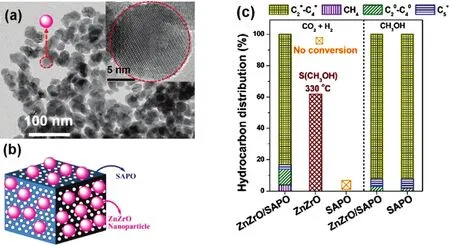
Fig. 9 (a) TEM and HRTEM (inset) images of ZnZrO. (b) Schematic description of ZnZrO/SAPO. (c) Catalytic results of CO2 hydrogenation on ZnZrO/SAPO, ZnZrO and SAPO, and methanol conversion on ZnZrO/SAPO and SAPO 41.Reproduced with permission from © 2017 American Chemical Society.

Table 3 Reported catalysts for the synthesis of high alcohols by CO2 hydrogenation.
In addition to composition, the structure of catalyst supports also influences catalytic activity and product selectivity.Ouyang et al.142synthesized two different morphologies of Co3O4(nanorods and nanoplates) and used them to support Pt NPs for CO2hydrogenation. The nanoplates were found to be reduced more easily than the nanorods, forming some metallic Co. The metallic Co had strong hydrogenation ability, and produced CH4dominantly. Whereas the Co3O4nanorods were reduced to CoO, which had a large mobility of surface oxygen.The surface oxygen was easily reduced to generate oxygen vacancies. The synergic effect of Pt and Co, and oxygen vacancies of reduced Co3O4 facilitated the production of high alcohols with C2+ alcohol selectivity of 22.7%. Bimodal MCM-41 (T) was revealed to be a better support for FeCu catalysts towards CO2hydrogenation than unimodal MCM-41(SS), yielding higher CO2conversion (1.2–2.1 times). The MCM-41 (T) catalyst loaded with 3% (w) Fe and 10% (w) Cu exhibited an alcohol (methanol and ethanol) selectivity of 80%–99% at low reaction temperature (160–200 °C). Such high activity was correlated with the pore characteristics of the support. The existence of larger mesopores in bimodal MCM-41 (T) was speculated to promote the formation of bigger metal particles, resulting in weaker metal-support interactions, beneficial to this reaction and yield of alcohols143.
5 Summary and outlook
Mitigating the effects caused by waste CO2emissions continues to be a critical issue to modern society. The hydrogenation of CO2by using renewable hydrogen offers an attractive approach for conversion of CO2into fuels or value-added chemicals. High (C2+) hydrocarbons and oxygenates are desirable hydrogenation products that bear advantages of high energy densities and compatibility with end-use infrastructures. However, the hydrogenation of CO2to yield C2+products remains an ongoing challenge due to high C―C coupling barriers.
CO2 can be hydrogenated to hydrocarbons via CO intermediate or CH3OH intermediate. The former involves an integration of RWGS reaction and FT-like synthesis. While the latter couples CH3OH synthesis with MTH process. The design of catalyst is the key to accomplishing the selective yield of C2+fuels. Advanced catalysts need to be active for both reductive step and C―C coupling step. ZnZrO/SAPO and In2O3/ZrO2/SAPO tandem catalysts gave the highest selectivity for lower olefins (80%–90%) via CH3OH intermediate to date.Fe or Co-based catalysts have been widely used to catalyze the hydrogenation of CO2to C2+hydrocarbons via CO intermediate. The incorporation of alkali metals and transition metals in Fe-based catalysts can enhance catalyst resistance to deactivation as well as CO2conversion to hydrocarbons. K appears to be the most effective alkali metal promoters,especially for the production of olefins. For Co-based catalysts,promoters play a role in suppressing methanation rate and improve selectivities to liquid hydrocarbons (C5+). FeCx(possibly Hägg iron carbide, X-Fe5C2) in Fe-based catalysts is considered to be the major active site for the synthesis of liquid hydrocarbons from CO2. Likewise, Co2C stabilized with a promoter (Mn) may be the active centers in FT synthesis88,although unpromoted Co2C is unstable in reaction conditions and is readily reduced to Co0. In addition to FeCx, Co2C, and Mo2C that have shown promising activities in FT synthesis,other metal or bimetal carbide catalysts deserve future exploration, which may give interesting catalytic properties for CO2conversion to C2+species.
High alcohols (mostly ethanol) have been produced by using a hybrid tandem catalyst. CO2activation occurs on Cu-based catalysts, noble metals or metal oxides with oxygen vacancies,whilst C―C coupling takes place within the pores of zeolites.The activation mechanism, C―C precise coupling, and synergy control between the two active components require further research.
In spite of these encouraging results, both the time-yields and selectivities of C2+products achieved so far are unsatisfactory. No commercial implementation has yet reached.In addition, the nature of active sites, and interactions among active phase, promoter, and support are still under debate.Developing in situ (spectroscopic) characterization techniques will help build more precise theoretical models to gain deeper insights into catalytic reaction pathways and structure-activity relationships. The combination of calculations and experiments will further aid catalyst design to increase the number of active sites and intrinsic activity of each active site. This also facilitates the development of novel robust catalysts with cooperative functionalities to achieve both high CO2conversion and C2+product selectivity.
杂志排行

物理化学学报的其它文章
- Catalytic Electroreduction of CO2 to C2H4 Using Cu2O Supported on 1-Octyl-3-methylimidazole Functionalized Graphite Sheets
- Green and Cost-Effective Preparation of Small-Sized ZSM-5
- Physicochemical Properties of1-Methoxyethyl-3-Methylimidazolium Glycine
- Study on Solution Enthalpies of Ionic Liquids [Cnmim][H2PO4](n = 3, 4, 5, 6) by Using Pitzer’s Equation
- Influence of External Electric Field on Vibrational Spectrum ofImidazolium-Based Ionic Liquids Probed by Molecular Dynamics Simulation
- Self-Assembly Behavior of Amphiphilic Diblock Copolymer PS-b-P4VP in CO2-Expanded Liquids