Influence of External Electric Field on Vibrational Spectrum ofImidazolium-Based Ionic Liquids Probed by Molecular Dynamics Simulation
2018-09-18CHENWenqiongGUANYongjiZHANGXiaopingDENGYouquan
CHEN Wenqiong , GUAN Yongji , ZHANG Xiaoping ,*, DENG Youquan
nstitute of Modern Communication, School of Information Science and Engineering, Lanzhou University, Lanzhou 730000,P. R. China.
entre for Green Chemistry and Catalysis, Lanzhou Institute of Chemical Physics, Chinese Academy Sciences, Lanzhou 730000,P. R. China.
Abstract: In this study, the influence of an external electric field (EEF) on the vibrational spectra of an imidazolium-based ionic liquid, 1-ethyl-3-methylimidazolium hexfluorophosphate (EMIMPF6), in the wavenumber range from 0 to 4000 cm−1 was probed by molecular dynamics (MD) simulation at 350 K.The results showed that the experimentally obtained spectrum could be reproduced by the calculated vibrational bands (VBs) in the wavenumber range from 400 to 4000 cm−1 using MD simulation without any EEF. When the EEF applied increased from 0 to 9 V·nm−1, the VB intensities at 50.0 and 199.8 cm−1 increased continuously and then tended to be saturated, while the VB intensities from 400 to 4000 cm−1 decrease and eventually disappear. Moreover, the VB at 50.0 cm−1 was red-shifted to ~16.7 cm−1 and then increased to 33.3 cm−1 as the EEF was increased from 0 to 2 and then to 3 V·nm−1 and higher. The VB at 3396.6 cm−1 was redshifted to ~16.7 cm−1 and then increased to 33.3 cm−1 as the EEF was increased from 0 to 3 and then to 4 V·nm−1 and higher; however, the position of other VBs from 0 to 4000 cm−1 remain almost unchanged. Based on further analysis of the simulation results and previously reported studies, for the VB at 50.0 cm−1, the increasing EEF enhances the polarity between cations and anions; thus, the difference in dipole moment between the cations and the anions increases, which continually increases the VB intensity until saturation is reached. For the VB at 199.8 cm−1, the increasing EEF intensifies the twisting of the ethyl chain, which enhances the VB intensity until saturation. For the other VBs from 400 to 4000 cm−1, the increasing EEF makes the orientation of the cations and anions in EMIMPF6 more consistent; thus, it can be conjectured that such consistent orientation may weaken the VB intensities and can even make them disappear. The redshift of VB at 50.0 cm−1 may occur because the EEF breaks the distribution of the electrostatic field inside EMIMPF6 and then weakens the interactions between cations and anions. The redshift of VB at 3396.6 cm−1 may be attributed to the EEF weakening the stretching vibration of the hydrogen bonds formed between the N atoms and the acidic hydrogen atoms on the cationic imidazolium rings. The EEF does not change the positions of the other VBs because the inherent stretching, bending, and rocking vibration of functional groups are not affected by the EEF.
Key Words: Vibrational spectrum; External electric field; Ionic liquid; Molecular dynamics simulation
1 Introduction
The vibrational spectrum (VS) is produced by the transition between different vibrational energy levels in the same electron energy state in the molecule, which is located in infrared region and is also known as infrared absorption spectrum. The VS is a multi-function tool to exhibit various conformational functional groups and hydrogen bonds of both solid and liquid materials and their solutions. Earlier research work on vibrational spectra mainly focused on water using experimental1and computational simulation2,3methods. Gan and coworkers1used the 10 Hz and 23 ps sum frequency generation (SFG)spectrometer laser system to process the experimental measurement data and presented a detailed analysis of the SFG vibrational spectra of the air/water interface considering different polarizations and experimental configurations.Praprotnik et al.3performed a classical molecular dynamics(MD) simulation with the flexible simple point charge (SPC)and flexible extended SPC (SPC/E) water models to investigate the influence of two temperatures of −4 and 80 °C on the VS.They found that the vibrational band (VB) associated with the bending mode is consistent with the experiment, so they confirmed that the MD simulation method can successfully reproduce all other experimentally observed spectroscopic properties of bulk water. Paesani et al.2investigated hydrogen bonds dynamics in liquid water with a specific focus on the relationship of these dynamics to VS using a novel simulation approach, which combines an ab initio-based force field for water with a quantum treatment of the nuclear motion. The calculated spectra are in good agreement with the available experimental data, indicating the accuracy of the present simulation approach in describing the properties of liquid water under ambient conditions.
Ionic liquids (ILs) composed of bulky organic cations and organic/inorganic anions have melting points near room temperature and are known as room temperature ILs (RTILs)4,5.RTILs have excellent physicochemical properties such as low volatilization, high chemical and thermal stability, high ionic conductivity, a large electro-chemical potential window and remarkable solubility6,7, which have been widely applied in lab on a chip8, variable-focus lenses9, flow-induced energy harvesting10,11, electro-wetting12,13in recent years. RTILs have attracted much theoretical interest for broad applications as environmentally friendly solvents in chemical and industrial processes, so a better understanding of physicochemical properties and structures of bulk pure RTILs is critically important for the vast majority of these applications. Jeon et al.14experimentally investigated the structures of ILs 1-butyl-3-methylimidazolium iodide, BMIMI and 1-butyl-3-methylimidazolium tetrafluoroborate, BMIMBF4and their aqueous mixtures from the attenuated total reflection (ATR)infrared absorption spectra. They found that the infrared absorption spectrum of cation BMIM+is very different in these two ILs and changes with increase of the water concentration due to the difference of the relative position between the anions and cations. Bhargava and Balasubramanian15studied the VS of imidazolium based IL 1,3-dimethylimidazolium chloride,MMIMCl which was calculated by the Fourier transform of the normalized velocity autocorrelation function (VACF) of all atoms obtained from the trajectory in the system using Car-Parrinello MD simulation (CPMD). They found that the results were in good agreement with spectroscopic data in the experiment, which indicated the hydrogen bond between the acidic hydrogen atom on the cationic imidazolium rings and the chloride ion could be obtained not only through structural data but also through vibrational dynamics of the cation and the latter was more direct comparing with experimental method.Katsyuba et al.16investigated possible molecular structure of the ion pairs in several imidazolium based ILs (halides,tetrafluoroborates, and hexafluorophosphate based imidazolium salts) using density functional theory (DFT) method in combination with VS, and meanwhile revealed both common features and differences in structure of various imidazolium based ILs. They also found that vibrations of cations not only depend on the conformational changes but also on the association with counterions on the basis of previous research.
As we all known, electric fields are significant to analyze the regulating macroscopic and microscopic properties of ILs.Ricks-Laskoski and Snow17pointed out that polymerizable ILs and their actuation in an electric field are combination of material and properties with unique potential to display structural and fluid dynamics above that found for small molecule ILs. However, there have few reports focused on the influence of the external electric field (EEF) on the VS for ILs using MD simulation method. Comparing with the DFT method, MD simulation method can study the VS for ILs systems containing more cation and anion pairs on the nanoscale. Under these circumstances, the VS for larger ILs systems under EEF need to be investigated using MD simulation method. In this study, we performed a classical MD simulation to investigate the influence of the EEF varied from 0 to 9 V·nm−1on the VS of imidazolium-based IL 1-ethyl-3-methylimidazolium hexfluorophosphate, EMIMPF6system consisting of 512 ILs pairs in the range from 0 to 4000 cm−1. The VS envelope was calculated by a Fourier transform of the normalized VACF. Firstly, we calculated the VS of EMIMPF6 in the range from 0 to 4000 cm−1and found the main features of the experimentally measured spectrum are reproduced by the calculated VBs using MD simulation.Secondly, the VS of EMIMPF6in the range from 0 to 4000 cm−1was calculated by a Fourier transform of the normalized VACF of all atoms obtained from the trajectory in the system under an EEF of 3 V·nm−1. Finally, the influence of EEF strength on the intensities and position of VS was also investigated. In particular, we found the EEF not only changes the intensities of the VS but also changes the position of the VS that make the VS show a redshift.
2 Computational methods
2.1 Molecular models
In this study, the imidazolium based IL EMIMPF6is employed to probe the VS under the action of the EEF varied from 0 to 9 V·nm−1using MD simulation, and the molecular structures of the cation and anion used are showed in Fig. 1.The initial cube model of EMIMPF6with dimensions of 5.16 ×5.16 × 5.16 nm constructed by PACKMOL software18, where the initial structure of the single IL pairs is derived from the Cambridge Crystallographic Data Centre (CCDC). Periodic boundary conditions (PBC) are applied to all directions of the simulated box in the whole MD simulation. Fig. 2 shows the ensemble structure with the initial cube model including 512 EMIMPF6ion pairs obtained from a separate MD simulation,which is visualized with a molecular graphics software named visual molecular dynamics (VMD)19.
2.2 Simulation details
All MD simulations were carried out using the general purpose parallel MD simulation open-source package DL_POLY 4.0820. The non-polarizable force field of ILs employed is based on the systematic all-atom force fields(AA-FFs) developed by the work of Canongia Lopes and Padua21that is in the frame of optimized potential for liquid simulation/all atom (OPLS-AA) force field22,23, which is a combination of parameters derived from quantum chemical calculations and force fields published in the literature. Its potential energy’s functional expression is

Fig. 1 The molecular structures of 1-ethyl-3-methylimidazolium(EMIM+) and hexafluorophosphate (PF6-).

Fig. 2 Initial structure of the ensemble.

where the total energy of the given system is the sum of bond stretch, bond angle bend, dihedral torsion, and pairwise nonbonding energies concluding the van der Waals (VDW)interactions computed by the Lorentz-Berthelot combining rules, and Coulombic interaction of partial atomic charges.
In the MD simulation process, the Newton’s motion equations were integrated by using a velocity verlet algorithm with an integration timestep of 2.0 fs. The initial velocities were assigned to the atoms randomly according to the system’s temperature. Long range Coulombic interactions were treated using the smooth particle mesh Ewald (PME) method24. The VDW and the real space electrostatic interactions were truncated and shifted to zero at 1.5 nm.
The MD simulation of EMIMPF6was employed with the system consisting of 512 ion pairs, and was performed using the Nosé-Hoover thermostat25,26. The thermostat was set to the temperature according to the melting point of EMIMPF6in the system. The initial configuration was then equilibrated with a NPT ensemble for p = 1.01325 × 105Pa and T = 350 K until the experimental value of the liquid density was achieved and the total energy of the system converged to a constant after 10 ns with time step of 2.0 fs (Fig. S1). Then the obtained equilibrated configuration at T = 350 K was allowed to run for another 10ns with a NVT ensemble to ensure a better equilibrium state. Finally, a sample procedure was taken every 1000 time steps for EMIMPF6configuration to obtain the statistically equilibrium averaged values in the next 10 ns.
2.3 Calculation of VS
The VS could be used to study structures and hydrogen bonds of ILs, and there are usually two methods to calculate the VS, which are the Fourier transforming of the system’s total dipole moment autocorrelation function and the Fourier transforming of the VACF3. In this paper, we calculate the VS using the second method. The VS is obtained by the Fourier transforming of the VACF as
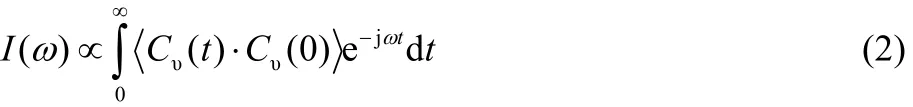
where I(ω) is the VS intensity, Cυ(t) is the centroid equivalence of the normalized VACFs of all atoms in the system at time t,ω is the VS frequency, and the angular bracket represents an average taken over all time origins. The normalized VACF of the single ion at time t is calculated by

where υc(t) is the velocity of the center of mass of the single ion in the system at time t. In strongly coupled molecular systems with a large number of atoms, the VACFs of all atoms should fluctuate and decay to zero very rapidly within a small number of vibrational periods about 2 ps.
3 Results and discussion
3.1 Without EEF
After the system reaches equilibration, the VS of imidazolium-based IL EMIMPF6is calculated by the Fourier transforming of the normalized VACFs of all atoms in the system. The VS for the pure EMIMPF6in the range from 0 to 4000 cm-1are showed in Fig. 3. It is clearly seen that the main features of the experimentally measured spectrum are reproduced by the calculated VBs using MD simulation (Fig.S2). Through comparing with the VS obtained from experiments27and DFT calculation16, we found the VS in Fig.3 which obtains from the MD simulation is reasonable and receivable. It is worth noting that the numbers of the VBs obtained using MD simulation are obviously less than the VBs measured in experiments, this because the EMIMPF6used in experiment is not completely pure IL and includes some impurities which can generate some noise peaks. Therefore, the VS obtained from MD simulation may more accurately reflect the real situation.
The VBs wavenumbers and assignments for EMIMPF6 are listed in Table 1. As showed in Table 1, the calculated vibrations assigned to these bands including C―H in the imidazolium ring stretching mode (3196.8 cm−1), the ring C=C stretching mode (1620.7 cm−1), the terminal CH3anti-symmetrical H―C―H bending mode (1465.2 cm−1), the terminal CH3 rocking mode (1132.2 cm−1), the ethyl side chain C―C stretching mode (949.0 cm−1), the ethyl side chain CH2and CH3rocking modes (782.6 cm−1), the N atom in the imidazolium ring and the ethyl side chain/terminal CH3rocking modes (432.9 cm−1), and the torsion of the ethyl side chain(199.8 cm−1). Particularly, there are two VBs at 50.0 cm−1assigned to the cations and anions torsion28–30belonging to far-infrared region and 3396.6 cm-1assigned to theintermolecular hydrogen bonds formed by N atoms and acidic H atoms on the cationic imidazolium rings. The VB at 50.0 cm−1can be attributed to the intermolecular interaction between cations and anions, which provides a new method to probe the cation-anion interaction in ILs by far-infrared spectrum30. To illustrate the association of the intermolecular hydrogen bonds,we calculate the radial distribution functions (RDFs) of the N atom and H2/H4/H5 atoms in the imidazolium ring. The RDFs for the N atom and the H atom in the imidazolium ring are showed in Fig. 4, which describes the probability of finding the selected atomic site around others in the system. From Fig. 4,we can get that the electrostatic interaction between N atom and H2 atom is stronger than between N atom and H4/H5 atom.The higher intensity of distribution between N atom and H2 atom in Fig. 4 indicates the intermolecular hydrogen bonds consisting of N atom and H2 atom in the imidazolium ring. To further illustrate the intermolecular hydrogen bonds between N atoms and H2 atoms, we calculate the average numbers of H2 atoms around N atoms in the radius of 0.3 nm and find that there are two H2 atoms around N atoms (Fig. S3). This can confirm the intermolecular hydrogen bonds are formed between N atoms and adjacent H2 atoms in different cations.

Fig. 3 The VS (arbitrary units) of EMIMPF6 calculated by Fourier transforming of all atoms’ VACFs without EEF.
3.2 EEF
The VS of EMIMPF6under an EEF of 3 V·nm−1in the range between 0 to 4000 cm−1are presented in Fig. 5. As showed in Fig. 5, the relative intensities of VBs at 50 cm−1and 200 cm−1significantly enhance under an EEF, while the VB intensities from 400 cm−1to 4000 cm−1obviously weaken under an EEF. On the other hand, the VBs at 50.0 cm−1and 3396.6 cm−1redshift about 33 and 16 wavenumbers respectively and the positions of other VBs are almost unchanged.
It is well known that the VBs intensities below 200 cm−1in the far-infrared region represent the interaction strength between cations and anions30, while the VBs between 400 cm−1and 4000 cm−1represent the vibrational characteristic of functional groups including groups on cationic imidazolium ring and ethyl/methyl side chains, and so on. Fig. 5 clearly shows that the VB intensities at 50.0 cm−1and 199.8 cm−1between cations and anions enhance under an EEF of 3 V·nm−1.In the case of the VB at 50.0 cm−1, the EEF makes the distance between cations and anions increase (from 0.528 to 0.534 nm)that enhances the polarity between cations and anions and thus the change of the dipole moment between the cations and the anions grows up. The increase of the change of the dipole moment makes the VB intensity at 50.0 cm−1enhance. In the case of VB at 199.8 cm−1, the EEF intensifies the twisting of the ethyl chain, which enhances the VB intensity at 199.8 cm−1.This is confirmed by the calculated the orientation of the ethyl chain in Fig. 6. From Fig. 6, we can find the values of second Legendre polynomials (P2(cos(θ)) = (3cos2(θ) − 1)/2) about the orientation angle significantly increase under an EEF of 3 V·nm−1comparing with the values without EEF. This indicates the twisting of the ethyl chain enhances indeed and so enhances the VB intensity at 199.8 cm−1. For other VBs from 400 cm−1to 4000 cm−1, the EEF changes the distribution structures of cations and anions and make them rearrange, thus the orientation of the cations and anions in EMIMPF6 is more consistent (Fig. 6) and we conjectured this consistent orientation maybe weaken the VB intensities.
As showed in Fig. 5, the EEF not only changes the VS intensities but also changes the VS position. Under an EEF of 3 V·nm−1, the VB at 50.0 cm−1redshifts about 33.3 wavenumbers. This redshift of VB at 50.0 cm−1is mainlycaused by the EEF breaks the distribution of the electrostatic field inside EMIMPF6and then weakens interactions between cations and anions. In order to more intuitively explain the weakening of the interactions between cations and anions, the RDFs of cations and anions are calculated and presented in Fig.7. It can be visually seen from Fig. 7 that the first peak value of RDFs under an EEF of 3 V·nm−1is less than the first peak value of RDFs without EEF. This indicates the interactions between cations and anions weaken under an EEF and thus the VB at 50.0 cm−1shows a redshift. Likewise, the VB at 3396.6 cm−1redshifts about 16.7 wavenumbers under an EEF of 3 V·nm−1. This redshift may be attributed to the EEF weakens the stretching vibration of hydrogen bonds formed by N atoms and acidic H atoms on the cationic imidazolium rings. The stretching vibration of hydrogen bonds weakening can be confirmed in results reported in Fig. 8. The first peak value of RDFs under an EEF of 3 V·nm−1is higher than the first peak value without EEF, which indicates the numbers of H atoms around N atoms increase, this can be confirmed by the calculated average numbers of H atoms around N atom which varies from 1.95 to 1.98 in the radius of 0.22 nm. Thus, the probability to form hydrogen bonds increases and more hydrogen bonds generate. Therefore, the stretching vibration of hydrogen bonds enhances and the VB at 3396.6 cm−1shows a redshift. For other VBs from 0 to 4000 cm−1, the EEF does not change the positions of VBs due to the inherent stretching,bending and rocking vibration of functional groups are not affected by the EEF.

Table 1 VS of EMIMPF6 without EEF.

Fig. 4 RDFs between nitrogen atom connecting the ethyl side chain and hydrogen atoms on the cationic imidazolium rings without EEF.

Fig. 5 The vibrational spectra (arbitrary units) of EMIMPF6 under an EEF of 0 and 3 V·nm-1.

Fig. 6 Average values of P2(cos(θ)) for the angles between the selected vector (N atom and thermal C atom on the ethyl side chain) and the vector that is perpendicular to the direction of EEF (z axis ) in the system under EEF of 0 and 3 V·nm-1.

Fig. 7 RDFs of cations and anions in EMIMPF6 under EEF of 0 and 3 V·nm-1.
Additionally, the influence of EEF strength on the VS of EMIMPF6was also investigated in the range from 0 to 4000 cm−1. The MD simulations performed with different EEF varied from 0 to 9 V·nm−1would allow for interference of the effect of EEF on the VS of EMIMPF6. Though applying different EEF on the EMIMPF6, we found the VB intensities at 50.0 cm−1and 199.8 cm−1continually enhanced and then tended to be saturated, while the VB intensities from 400 cm−1to 4000 cm−1obviously weakened and eventually disappeared with increase of EEF strength (Fig. S4). The VS of EMIMPF6under the EEF varied from 0 to 4 V·nm−1were showed in Fig.9. For the VB at 50.0 cm−1, the increasing EEF enhances the polarity between cations and anions and thus the change of the dipole moment between the cations and the anions grows up,which makes the VB intensity continually enhance and then reach saturation. For the VB at 199.8 cm−1, the increasing EEF intensifies the twisting of the ethyl chain, which makes the VB intensity increase to be saturated. For other VBs from 400 to 4000 cm−1, the increasing EEF makes the orientation of the cations and anions in EMIMPF6more and more consistent and we conjectured this consistent orientation maybe weaken the VB intensities and even make them disappear. Moreover, as showed in Fig. 10, the VBs at 50.0 cm−1redshifted about 16.7 cm−1as the EEF varied from 0 to 2 V·nm−1and the redshift increased to 33.3 cm−1as the EEF varied from 2 to 3 V·nm−1and further > 3 V·nm−1. The VBs at 3396.6 cm−1redshifted about 16.7 cm−1under the EEF varied from 0 to 3 V·nm−1and the redshift increased to 33.3 cm−1as the EEF varied from 3 to 4 V·nm−1and further > 4 V·nm−1, but the position of other VBs from 0 to 4000 cm−1are almost unchanged. The redshift of VB at 50.0 cm−1from 16.7 to 33.3 cm−1may be caused by the increasing EEF first breaks the distribution of the electrostatic field inside EMIMPF6and eventually keep balance with the inner electrostatic field. The redshift of VB at 3396.6 cm−1is not entirely clear and this result may be attributed to the increasing EEF weakens the stretching vibration of hydrogen bonds formed by N atoms and the acidic hydrogen atoms on the cationic imidazolium rings and finally the stretching vibration is almost unchanged. For other VBs, the EEF does not change the positions of VBs due to the inherent stretching, bending and rocking vibration of functional groups are not affected by the EEF.

Fig. 8 RDFs between N atom connecting the ethyl side chain and H atoms on the cationic imidazolium rings under EEF of 0 and 3 V·nm-1.

Fig. 9 The vibrational spectra (arbitrary units) of EMIMPF6 under different EEF.
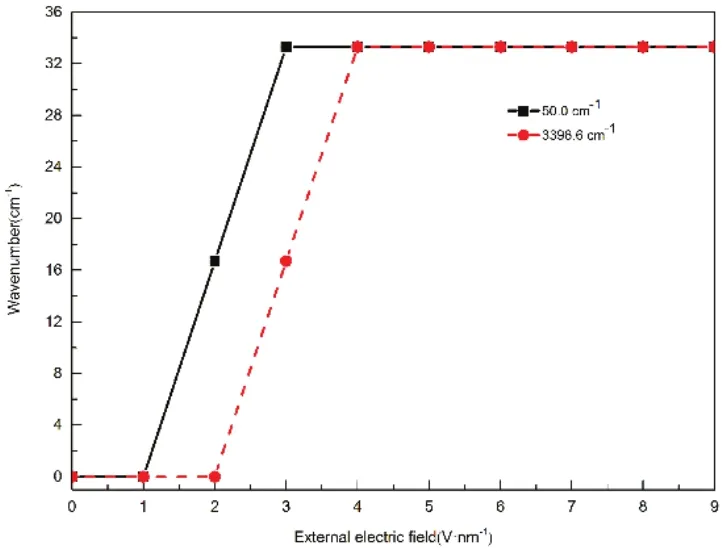
Fig. 10 The redshift of VBs at 50.0 and 3396.6 cm-1 under different EEF.
4 Conclusions
In summary, here we investigate the influence of EEF on the vibrational spectra of imidazolium-based IL EMIMPF6in the range from 0 to 4000 cm−1using classical MD simulation. The research results showed that the experimentally measured spectrum could be reproduced by the calculated VBs in the range from 400 to 4000 cm−1using MD simulation without such EEF. Under the EEF ranging from 0 to 9 V·nm−1, the VB intensities at 50.0 and 199.8 cm−1continually enhance then tend to be saturated, while the VB intensities from 400 to 4000 cm-1obviously weaken and eventually disappear. Moreover, the VB at 50.0 cm−1redshifted about 16.7 then increased to 33.3 cm−1as the EEF varied from 0 to 2 then to 3 V·nm−1and further. The VB at 3396.6 cm−1redshifted about 16.7 then increased to 33.3 cm−1as the EEF varied from 0 to 3 then to 4 V·nm−1and further, but the position of other VBs from 0 to 4000 cm−1are almost unchanged. Based on further analysis on the simulation results and previous reported literatures, for the VB at 50.0 cm−1, the increasing EEF enhances the polarity between cations and anions and thus the change of the dipole moment between the cations and the anions grows up, which makes the VB intensity continually enhance and then reach saturation. For the VB at 199.8 cm−1, the increasing EEF intensifies the twisting of the ethyl chain, which makes the VB intensity increase to be saturated. For other VBs from 400 to 4000 cm−1, the increasing EEF makes the orientation of the cations and anions in EMIMPF6 more consistent and it can be conjectured that such more consistent orientation may weaken the VB intensities and even make them disappear. The redshift of VB at 50.0 cm−1may be caused by the EEF breaks the distribution of the electrostatic field inside EMIMPF6and then weakens interactions between cations and anions. The redshift of VB at 3396.6 cm−1may be attributed to the EEF weakens the stretching vibration of hydrogen bonds formed by N atoms and the acidic hydrogen atoms on the cationic imidazolium rings.For other VBs, the EEF does not change the positions of VBs due to the inherent stretching, bending and rocking vibration of functional groups are not affected by the EEF. These results may be significant for opening prospects to investigate the effect of external field on properties of ILs.
Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn.
杂志排行

物理化学学报的其它文章
- BmmimOAc-Catalyzed Direct Condensation of 2-(Arylamino) Alcohols to Synthesize 3-Arylthiazolidine-2-thiones
- Self-Assembly Behavior of Amphiphilic Diblock Copolymer PS-b-P4VP in CO2-Expanded Liquids
- Catalytic Electroreduction of CO2 to C2H4 Using Cu2O Supported on 1-Octyl-3-methylimidazole Functionalized Graphite Sheets
- Study on Solution Enthalpies of Ionic Liquids [Cnmim][H2PO4](n = 3, 4, 5, 6) by Using Pitzer’s Equation
- Physicochemical Properties of1-Methoxyethyl-3-Methylimidazolium Glycine
- Green and Cost-Effective Preparation of Small-Sized ZSM-5