H,Cl和F原子钝化Cu2ZnSnS∗4(112)表面态的第一性原理计算
2018-09-11王小卡汤富领薛红涛司凤娟祁荣斐刘静波
王小卡 汤富领† 薛红涛 司凤娟 祁荣斐 刘静波
1)(兰州理工大学,材料科学与工程学院,省部共建有色金属先进加工与再利用国家重点实验室,兰州 730050)
2)(德克萨斯农工大学化学系,美国德克萨斯州金斯维尔市大学大道700号,美国 78363)
1 引 言
铜铟镓硒(CuInGaSe2,CIGS)基薄膜太阳能电池的技术已经成熟,实现了商业化生产,其转换效率达到了21.7%[1].然而In和Ga稀贵,限制了该类电池的发展.四元化合物Cu2ZnSnS4(CZTS)与CIGS化合物具有相似的结构特性,有良好的光电性质,属于直接禁带半导体,带隙宽度可达到1.5 eV(1.05—1.5 eV),与太阳能电池最佳带隙宽1.5 eV相匹配,吸收系数超过104cm−1[2−7].同时,其成本低,原料丰富以及无毒,近几年备受关注[8].CZTS作为薄膜太阳能电池吸收材料,有非常好的发展前景,被认为是最有可能替代昂贵CIGS的材料之一.Cu2ZnSnS4基太阳能电池的光电转换效率目前已经提高到近12.6%[9,10].然而,它仍远低于32%的理论转换效率[11,12]和CIGS太阳能电池21.7%的转换效率.CZTS太阳能电池具有类似于CIGS太阳能电池的叠层状结构:前电极|ZnO窗口层|CdS缓冲层|CZTS吸光层|Mo背电极|玻璃.电池中各层材料的表面[13]和界面[14]性质对电池性能有重要的影响.电池的性能不仅取决于各层材料的结构和性能,还取决于每一层表面的处理和状态.因此,有必要从理论和实验上研究CZTS吸收层的表面结构和性质,探索表面对CZTS太阳能电池性能的可能影响.
近几年,研究者针对CZTS表面处理进行了试验研究,并取得一些很有意义的成果.Furuta等[15]在CdS缓冲层沉积之前对于沉积的Cu2ZnSn(S,Se)4(CZTSSe)薄膜先后进行HCl刻蚀和硫脲处理,这种连续处理方法对其表面起到了钝化作用.CZTSSe太阳能电池通过空气退火后,其转换效率达到10.5%.Lee等[16]通过原子层沉积生长Al2O3层改善了CZTS太阳能电池的开路电压、填充因子、长波长收集效率和短路电流密度器件,从而提高了功率转换效率.其方法为:沉积Al2O3薄膜在CZTS吸收层上,并且用原子层沉积生长的Al2O3薄膜代替叠层中的ZnO层(在缓冲层顶部),以减轻由该层引起的对CdS表面的损伤.Lin等[17]发现通过操纵双面处理中的钠含量,可以实现缺陷钝化并有效减少载流子复合,从而使CZTS器件性能从4.1%提高到5.6%.
理论计算是深入探索半导体材料表面、界面状态的有效途径.Xing等[18]通过第一性原理计算研究了H2S吸附在6 H-SiC(0001)表面上钝化的差异性和稳定性.Ohno和Shiraishi[19]研究了S单层吸附的Ga和As终端的GaAs(001)表面,在带隙内的表面态密度明显降低.Wang等[20]利用第一性原理研究了H,Cl,F,GaO和S与氧化的GaAs(001)表面相互作用.H,Cl,F和GaO可以完全清除间隙区域表面态,因此成为良好的钝化候选物.Medaboina等[21]研究表明悬挂键表面钝化对纳米线电子结构产生了实质性影响,这种影响可能在各种传感和纳米电子应用中都是有益的.
光生载流子复合率与表面态成正比,电池材料表面或界面处的载流子复合会降低太阳能电池的光电转换效率,这可以通过在背表面引入一个纳米钝化层来实现对电池的优化[22,23].对于CZTS薄膜太阳能电池表面和界面的理论研究方面,Cheng等[24]观察到F,Cl和H原子钝化后CZTS(102)/WZ-ZnO(110)界面态的峰值减少.Huang等[25]从实验和理论上证明了简单的表面改性可以显着改变纳米晶薄膜中的载流子传输特性.Xu等[8]利用第一性原理计算方法分析了CZTS(112)/(¯1¯1¯2)表面模型的电子结构,发现富 Cu表面在带隙中产生有害的态密度,而Cu耗尽的表面没有表面态,此结果佐证了缺Cu和富Zn的CZTS太阳能电池可以具有更高的效率.
上述研究尽管给出了CZTS表面的一些实验和计算结果,但是还没有对表面原子吸附结构及钝化表面态机理展开详细的理论研究.因此,对表面态钝化进行深入的理论研究是必要的.本文通过第一性原理计算研究了H,Cl和F对CZTS(112)表面态的影响(CZTS(112)面为择优生长面).为确定最稳定的表面吸附构型,计算了原子的吸附能,同时分析了最稳定构型的晶体结构、键长和电子性质.
2 计算方法
理论计算工作采用基于密度泛函理论(density functional theory,DFT)的维也纳从头计算软件包(Vienna ab initio simulation package,VASP)[26−28]完成.电子交换关联泛函选取常用的广义梯度近似(general gradient approximation,GGA)中的Perdew-Burke-Ernzerhof(PBE)泛函[29,30].采用由Blöchl校正调节的四面体方法[31]计算在体相和表面的总能量,能带结构和态密度(density of states,DOS).离子弛豫采用共轭梯度(conjugate gradient)方法[32].由于使用DFTPBE泛函计算的半导体带隙通常很低,所以我们用GGA+U方法进行计算以得到较准确的电子性质[33,34],实际计算过程可通过设置库仑作用参数U和交换关联参数J之间的差值(Ueff=U−J)来修正未满态d轨道电子的强关联作用[35,36].我们使用的Cu-3d,Zn-3d和Sn-4d的U值分别为:5.2,6.3和3.3 eV,J值都为0 eV.这些与以前研究的U值校正一致[5,37].电子与离子芯之间的相互作用采用投影缀加平面波方法(projector augmented wave)[38,39]来描述.铜、锌、锡和硫的电子构型分别为[Ar]3d104s1,[Ar]3d104s2,[Kr]4d105s25p2和[Ne]3s23p4.对于CZTS体相和CZTS(112)表面的计算,所使用的平面波截断能全部为450 eV,k网格分别为5×5×2和6×8×1.
3 结果与讨论
3.1 CZTS体相的结构和电子性质
自然界中CZTS有锌黄锡矿和锌锡矿两种结构,锌黄锡矿结构的CZTS比锌锡矿结构的CZTS更稳定[2,5].CZTS属于I2-II-IV-VI4的四元硫属化物半导体(I=Cu,Ag;II=Zn,Cd;IV=Si,Ge,Sn;VI=S,Se)系列.锌黄锡矿结构的空间群为I¯4mc,结构如图1所示.CZTS优化的晶格常数是a=5.447 Å和c=10.854 Å. 实验值为a=5.427 Å和c=10.871 Å[40],采用DFT方法的计算值为a=5.477 Å,c=10.941 Å[41].与其他实验和计算结果非常接近,因此我们计算的晶格常数是可靠的.
CZTS体相能带结构和态密度如图2所示.图2(a)显示CZTS是一种直接带隙半导体材料.我们通过GGA+U方法计算其带隙宽度为0.84 eV,小于其他实验获得的带隙宽度1.4—1.6 eV[2,3,5−7,9],使用GGA方法和使用HSE06计算得到带隙宽度分别为0.09 eV[42]与1.3 eV[8],而CZTS理论带隙宽度为1.05—1.5 eV[43].CZTS体相的态密度计算结果如图2(b)所示,可以看到CZTS体相的价带顶部主要来自Cu-3d和S-3p轨道电子的贡献,而导带底部主要来自Sn-5s轨道电子的贡献.将我们的结果与其他研究进行比较[44,45],我们的计算结果与其相符.

图1 CZTS体相的晶体结构Fig.1.Crystal structure of bulk CZTS.

图2 CZTS体相的能带结构(a)和态密度(b)Fig.2.Band structure(a)and DOS(b)of bulk CZTS.
3.2 CZTS(¯1¯1¯2)/(112)表 面 的 结 构 和 电子性质
以金属原子 Cu-Zn-Sn 为终止的 CZTS(¯1¯1¯2)和以S原子为终止的CZTS(112)表面在弛豫前的晶格模型分别如图3(a)和图3(b).我们都选取了六个双原子层的表面模型,为了避免平板上下两个表面之间电荷转移,采用赝氢原子钝化平板底部表面上的悬挂键.同时在表面体系中添加了约6个原子层厚度的真空层以消除周期性边界对表面的影响. 我们分别对CZTS(¯1¯1¯2)/(112) 表面附近的三层结构进行弛豫,远离表面层的底部三层结构进行固定.通过分析层间距,键长以及局域态密度验证了六层结构足够模拟表面性质.

图 3 Cu-Zn-Sn 终止 CZTS(¯1¯1¯2)表面模型 (a)和 S 原子终止CZTS(112)表面模型(b)Fig.3.Cu-Zn-Sn-terminated CZTS(¯1¯1¯2)surface model(a)and S-terminated CZTS(112)surface model(b).
图4(a)和图4(b)分别为弛豫前后Cu-Zn-Sn终止的 CZTS(¯1¯1¯2)表面晶格结构. 对比图 4 中弛豫前与弛豫后结构,发现表面layer 1中金属原子在弛豫后没有旧键断裂或者新键形成,但Cu原子和Zn原子明显向表面内收缩,Sn原子向外伸展,S原子位置几乎没有变化,最终Cu,Zn与S原子在同一层.从图4(b)中可以看出,layer 1至layer 5中相邻两层层间距依次为2.30,2.25,2.32和2.31 Å;layer 2,layer 3以及layer 4(固定层)内金属原子和S原子之间的距离分别为0.62,0.68和0.68 Å.layer 2开始键长和层间距的变化很小,并且layer 3原子的键长与体相中相应键长非常接近.图4(c)所示layer 3的局域态密度(local density of states,LDOS)与体相最接近,费米能级附近几乎没有表面态. 因此, 用六个双原子层模拟CZTS(¯1¯1¯2)表面的性质是足够厚的.与layer 3相比,在layer 1中费米能级(0 eV)附近的表面态完全消失,在0.7 eV附近的表面态也有明显降低,可能是因为金属终止的CZTS(¯1¯1¯2)表面重构, 即自钝化. 在重构过程中金属原子位置变化,导致layer 1中原子之间的键长也发生了明显的变化,部分键长如图4(b)所示.重构后layer 1内Cu—S,Zn—S和Sn—S的键长分别为:2.21—2.26,2.24—2.31,2.58—2.60 Å.Cu—S和Zn—S键长比弛豫之前分别缩短了0.04—0.09 Å和0.04—0.11 Å,Sn—S键长增长了0.13—0.15 Å.layer 2之后的键长变化不大.

图4 CZTS(¯1¯1¯2)表面弛豫前(a)和弛豫后模型(b);弛豫后第三层(c)和第一层(d)的局域态密度Fig.4.CZTS(¯1¯1¯2)surface model before(a)and after(b)relaxation;LDOS of layer 3(c)and layer 1(d)after relaxation.
3.3 H,Cl和F原子吸附S原子终止的CZTS(112)表面
S原子终止的CZTS(112)表面结构弛豫前后结构没有明显变化,表1列出了弛豫后前三层原子的键长变化百分比.由表1中可知,层间键长比层内的键长变化明显,layer 1内键长变化比较大,键长变化率最大的是∆dZn—S(layer 1-layer 2),达到了4.68%.在layer 2开始键长变化已经很小(最大变化率∆dZn—S为1.66%),layer 3结构已经非常接近体相(最大变化率∆dZn—S为0.68%).说明六层表面模型模拟CZTS(112)表面是足够厚的.图5所示为CZTS(112)表面的总态密度(total density of states,TDOS)(a)、layer 1(b)和layer 4(c)的局域态密度.CZTS(112)表面总的态密度在费米能级0—0.3 eV附近有明显的态密度峰.对比CZTS(112)表面layer 1与layer 4的局域态密度,我们发现费米能级附近的态密度主要是由layer 1原子引起的,并且layer 4的局域态密度与体相的态密度最接近,进一步说明六层原子模型足够模拟该表面性质.为了更深入地了解其表面的电子性质,我们进一步分析了layer 1原子的分波态密度(partial density of states,PDOS).由图5(d)—(g)在费米能级0—0.3 eV附近的态密度可知,其表面态主要是由Cu-3d和S-3p轨道杂化引起的,在费米能级1.7—2.2 eV附近态密度主要是由Sn-5s轨道电子引起.对比体相中(图2(b))导带底部和价带顶部主要分布着Cu-3d,S-3p和Sn-5s轨道,说明表面原子的突然终止使得带隙附近的电子轨道发生偏移,进而在表面产生不利的表面态.
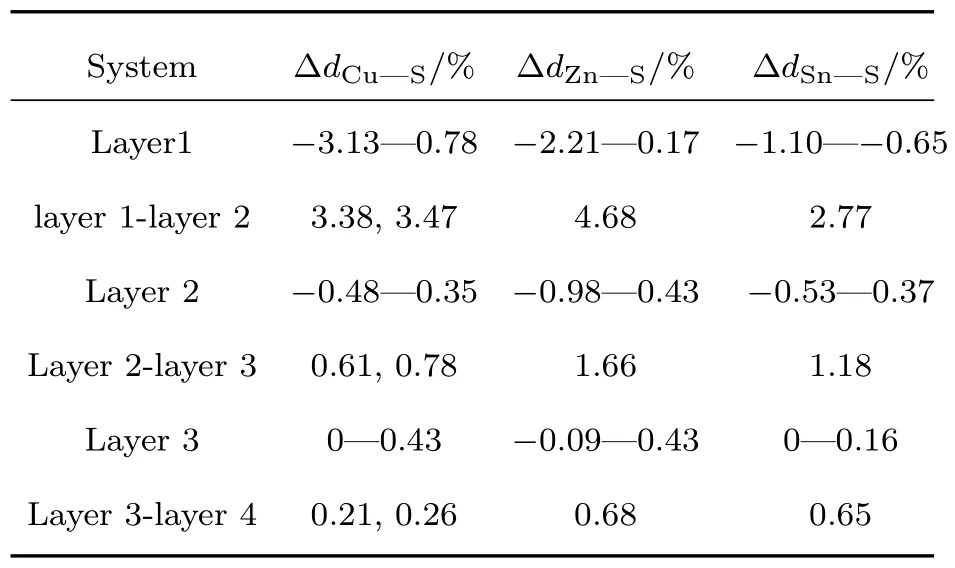
表1 CZTS(112)表面结构弛豫后的键长变化率Table 1.Change rate of bond length after CZTS(112)surface relaxation.
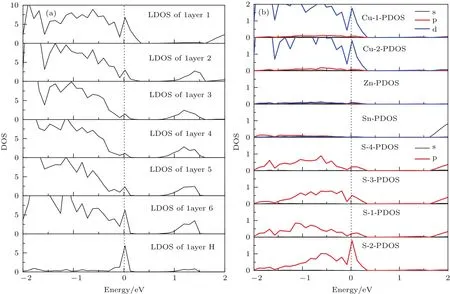
图5 CZTS(112)表面模型的局域态密度(a),第一层原子的分波态密度(b)Fig.5.LDOS of CZTS(112)surface(a),PDOS of atoms in layer 1(b).
在金属原子 Cu-Zn-Sn 终止的 CZTS(¯1¯1¯2)表面重构改变了电子结构并自钝化表面态.S原子终止CZTS(112)表面在费米能级附近有明显的表面态,尝试H,Cl和F原子吸附在其表面来钝化表面态.当覆盖度0.25 ML(此覆盖度为表面吸附原子数与表面S原子数比值)H,Cl和F原子吸附在CZTS(112)表面上时,我们研究了四种可能的吸附位点(如图6所示):top,bridge,hcp(位于第二层金属原子的顶部)和fcc(位于第四层金属原子的顶部)位点.由于H和Cl原子的四个高对称吸附位与F原子的对称吸附位相同,因此在图6中我们只给出了F原子的吸附位置.有四种类型的吸附位点,图6(a)代表四个top位点1—4,图6(b)代表八个bridge位点a—h,图6(c)代表四个hcp位点h-Cu1,h-Cu2,h-Zn,h-Sn和四个fcc位点f-Cu1,f-Cu2,f-Zn,f-Sn.我们计算了各位点的吸附能,其定义如下[46−48]:

根据(1)式,本文计算了三种原子在图6所示的各吸附位点上的吸附能,其吸附能较低的位置以及对应的吸附能如表2所列.表2中三种吸附原子优化后几乎都稳定吸附在top2位,只有H原子吸附在f-Zn位时,优化后移动至top2位正下方的S原子与h-Cu1位点之间的位置.从表2可以看出,H,Cl和F原子最低吸附能为−3.188,−2.279,−3.395 eV,单个H原子在f-Zn的吸附能与其他位置相比明显偏大.三种原子吸附最稳定的结构,其表面态没有明显的降低.考虑到吸附能量和态密度降低两个方面,我们继续计算了覆盖度为0.5 ML时(即表面添加两个同种吸附原子)三种原子在CZTS(112)表面的吸附情形.吸附位置的选择是利用覆盖度0.25 ML时表2吸附能较低的几个位置进行两两组合,其组合吸附位置以及对应的吸附能列于表3.

图6 CZTS(112)表面四种吸附位置:top(a),bridge(b),fcc和hcp(c)Fig.6.Four adsorption sites on the CZTS(112)surface:top(a),bridge(b),and fcc and hcp(c)sites.

表2 覆盖度为0.25 ML时原子的吸附能Table 2.Atom adsorption energy at 0.25 ML coverage.
当覆盖度为0.5 ML时,优化后H和Cl原子(H和Cl原子优化前的位置组合分别为:f-Cu1,f-Zn;2,f-Sn)分别吸附在top2和top3位点(图7(a)和图7(b))处具有最低的吸附能,其吸附能分别为−2.867和−2.020 eV(表3).在F原子的位置组合中,h-Zn,f-Sn吸附时能量最低(−3.132 eV),优化后的f-Sn位点F原子与top2下方的S原子键合(S-2),h-Zn位点F原子与其正下方第一层的Zn原子键合,并且Zn原子向外伸展.这可能是由于F原子的电负性很强,优化前位置偏低,容易与第一层金属结合.这就使得表面晶格结构发生很大变化,不利于表面态的钝化.
考虑到覆盖度为0.25 ML时,原子稳定吸附在top2位,且覆盖度为0.5 ML时H和Cl原子稳定吸附在top2和top3位,我们尝试在top2和top3位同时添加F原子.优化后发现原子仍在顶位(图7(d)),并且其吸附能为−3.141 eV,是覆盖度0.5 ML的F原子位置组合中是最低的(表3).因此我们认为top2和top3位点的组合是最稳定的吸附结构.对于不同元素的稳定吸附结构,H原子在S终止的CZTS(112)表面的吸附能为−2.867 eV,小于Cl原子钝化的吸附能(−2.020 eV),F原子的吸附能(−3.141 eV)最低.说明用F原子钝化后的体系最稳定,H原子钝化后的体系比Cl原子更稳定.如图7 Front view(a),图7(b)和图7(d)所示,三种吸附原子稳定吸附在top2和top3位,吸附原子与表面S原子结合成键.H原子相对于表面S原子的高度分别为1.319 Å和1.363 Å,dH—S键长为1.376 Å.Cl原子相对于表面S原子的高度分别为2.067 Å,dCl—S键长为2.079 Å.F原子相对于表面S原子的高度分别为1.671 Å和1.637 Å,dF—S键长为1.660 Å与1.668 Å.其表面原子吸附并非吸附于S原子的正上方,并且其原子偏离S-2和S-3的位置都相近,如图7 Top view(a),图7(b)和图7(d)所示(S-1,S-2,S-3,S-4为表面的四个S原子,其位于吸附位点top1,top2,top3和top4对应的正下方).

表3 覆盖度为0.5 ML时原子的吸附能Table 3.Adsorption energy at 0.5 ML coverage.

图7 覆盖度0.5 ML时,H(位置组合为f-Cu1,f-Zn)(a)、Cl(位置组合为top2,f-Sn)(b)、F(位置组合为h-Zn,f-Sn)(c)和F在两个顶位(位置组合为top2和top3)(d)吸附CZTS(112)表面时弛豫后的结构Fig.7.At 0.5 ML coverage,H(f-Cu1,f-Zn)(a),Cl(top2,f-Sn)(b),F(h-Zn,f-Sn)(c)and F(top2,top3)(d)adsorption on CZTS(112)surface structure after relaxation.
对于图7(a),图7(b)和图7(d)所示的三种最稳定构型,它们在费米能级附近的态密度与纯净表面的相比有明显的衰减.由于主要是前两层受表面悬挂键的影响产生缺陷能级,因此本文将前三层原子的局域态密度分别比较来分析其表面的钝化效果,它们的态密度图如图8所示.从费米能级处态密度峰值降低与带隙宽度的增加看出三种原子钝化效果都很明显,在H和Cl钝化后layer 1费米能级处态密度峰值几乎降为0,其带隙宽度从钝化之前的0.319 eV分别增大至0.525 eV和0.423 eV,F钝化后态密度峰值分别降低到原来的约1/3,其带隙为0.348 eV.layer 2和layer 3态密度峰值很小,添加钝化原子后也发生不同程度的减少.由于表面态影响表面与界面载流子的传输,所以钝化之后表面态减弱,这对太阳能电池表面载流子传输有利.
为了进一步探究表面态钝化原理,我们计算了三种吸附原子和表面原子的分波态密度,差分电荷密度和拜德电荷[49].三种原子吸附原子和表面layer 1内原子的分波态密度,如图9所示.H和吸附后表面与纯净表面相比,Cu-3d原子和S-3p原子在费米能级处的峰值几乎完全消失,F钝化表面上Cu-3d原子和S-3p原子在费米能级处的峰值明显降低,但是明显高于H和Cl原子,同时三种原子在费米能级附近没有引起新的表面态.钝化后Zn原子和Sn原子在费米能级处表面态更干净.为了分析钝化表面态的原因,对其差分电荷密度进行分析,其公式如下:

其中ρCZTS-x为整个吸附体系的电荷密度,ρCZTS和ρx为在同一体系中单独的CZTS(112)表面和吸附原子的电荷密度.如图10所示(图中黄色区域代表正值,意味着得到电子;绿色区域代表负值,意味着失去电子),结合原子的分波态密度和拜德电荷,分析其钝化机理和差异.如表4所列,钝化后S-2和S-3原子的拜德电荷相比钝化之前都是失去电子的,而Cu-1和Cu-2原子(h-Cu1和h-Cu2正下方)都得到少量电子(约0.09 e),更加接近体相的拜德电荷,吸附原子都是得到电子.所以我们推测添加吸附原子后,吸附原子从S原子得到电子,消除了部分悬挂键(结合表面S原子的分波态密度图中费米能级附近电子几乎完全消失,我们推测吸附原子可能从表面S-2和S-3原子得到了引起表面态的电子),表面电荷重新分配,使得内部Cu-1和Cu-2原子的电子轨道发生偏移,费米能级附近态密度峰降低.分析三种吸附原子的拜德电荷可以发现,钝化效果最好的H和Cl原子,在S-3原子顶位吸附时,得到的电子分别为0.01 e和0.20 e,而F原子得到了0.97 e.根据图中S-3原子的分波态密度图可知,其表面态相比S-2原子的峰值低很多.所以我们推测F原子从S-3原子得到了较多的电子,对于表面态的钝化是不利的.

图8 CZTS(112)表面钝化前后上三层的局域态密度Fig.8.LDOS of upper three layers before and after CZTS(112)surface passivation.
分析上述结果,我们发现S-2,S-3,Cu-1和Cu-2是导致表面态的主要原子;通过H,Cl和F吸附表面能量的测试,发现吸附在S-2和S-3原子顶部时最稳定,钝化效果明显,因此S-2和S-3原子是表面钝化的主要对象.H原子钝化效果略好于Cl原子,Cl原子比F好.根据S-3原子费米能级处的峰值相比S-2原子弱很多,我们推测F原子电负性强会从S-3得到过多的电子,导致电子重新分配后其钝化效果比H和Cl原子的效果稍差.

图9 H(a),Cl(b)和F(c)原子稳定吸附在CZTS(112)表面第一层原子的分波态密度Fig.9.PDOS of atoms in CZTS(112)layer1 with H(a),Cl(b)and F(c)stable adsorption.
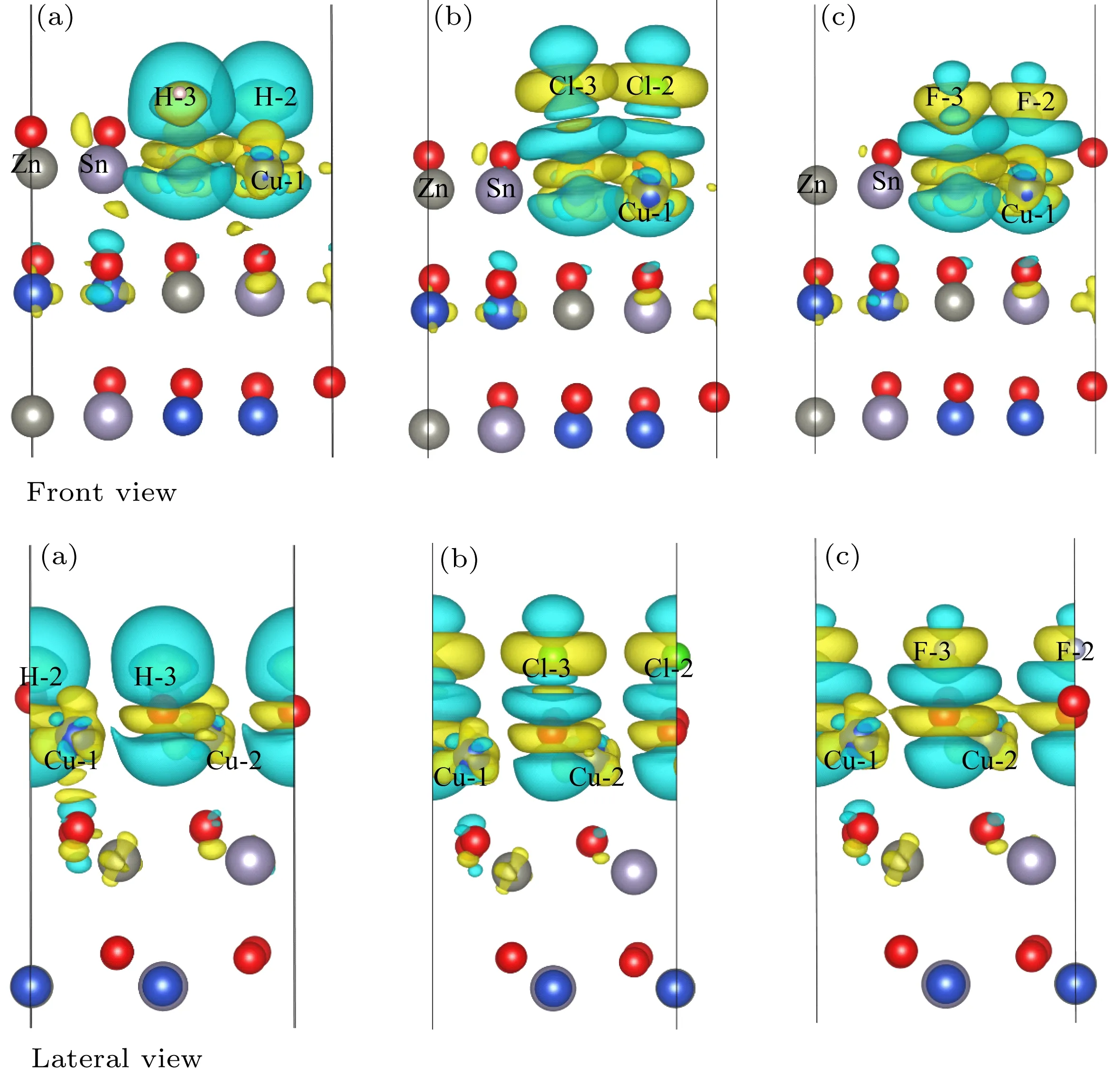
图10 H(a),Cl(b)和F(c)原子稳定吸附CZTS(112)表面后的差分电荷Fig.10.The charge density difference of CZTS(112)surface with H(a),Cl(b)and F(c)stable adsorption.
4 结 论
通过计算我们发现金属原子Cu-Zn-Sn终止的Cu2ZnSnS4()表 面 会 发 生 重 构 和 自 钝 化. 对于H,Cl和F原子吸附S终止的Cu2ZnSnS4(112)表面,计算发现覆盖度0.5 ML时原子稳定吸附在表面态明显的两个S原子的顶位.对于覆盖度0.5 ML H,Cl和F吸附最稳定的结构,三种吸附原子通过从两个特定的S原子获得电子,使表面电子重新分配,表面态得到钝化,理论上有助于光生载流子的传输.两个S原子为CZTS(112)表面态钝化的关键.同时,其钝化效果为:H>Cl>F,H和Cl原子钝化效果明显比F原子好.根据拜德电荷的差异,推测这与F原子从S-3原子获得较多的电子有关.
本工作完成于甘肃省超算中心.
