纤维诱导对乳清浓缩蛋白起泡性的改善作用分析
2018-08-31徐红华谭俊艳谢明明
徐红华 谭俊艳 谢明明 丁 瑞 关 琛 王 欣
(东北农业大学食品学院, 哈尔滨 150030)
0 引言
乳清蛋白经常作为发泡剂应用于蛋白糖饼、蛋糕、人造奶油和发酵面包等发泡性食品中。在低pH值(小于2.5)、低离子强度、高温长时间加热条件下,许多蛋白质能够自组装形成长的、有分支/无分支、半柔性淀粉样纤维[1],如乳清浓缩蛋白(WPC)[2-3]、β-乳球蛋白[4-5]、大豆蛋白[6]、牛血清白蛋白[7]和溶菌酶[8]。由于其较高的纵横比(长度与直径比值),纤维状聚合物可被用作增稠剂、胶凝剂[9-10]和乳化稳定剂[11-12]。在众多的性质中,蛋白纤维聚合物起泡性与泡沫稳定性远优于蛋白质的常规聚合物[13]。近年来研究发现,成熟纤维还可以促进蛋白质纤维化作用[14]。KREBS等[15]发现了成熟纤维加入到蛋白溶液中可加速形成母鸡溶菌酶纤维,并形成无分枝的致密网状结构。然而,关于纤维诱导蛋白成纤维的研究主要集中在聚合机理以及形成条件上,缺乏功能性质的研究。而纤维诱导蛋白形成的纤维化作用与其非诱导情况下自发形成的纤维作用,在功能方面是否存在差异尚缺乏研究。因此,本文采用乳清浓缩蛋白中加入一定量的成熟纤维,比较纤维诱导乳清浓缩蛋白成纤维与乳清浓缩蛋白自发形成纤维过程中起泡性的差异,以期为乳清浓缩蛋白纳米纤维的应用提供新的参考。
1 材料与方法
1.1 材料与仪器
主要材料有:乳清浓缩蛋白WPC-80(美国HILMA公司);硫黄素 T、8-苯氨基-1-萘磺酸(ANS)(Sigma公司);DTNB(Merck公司);其他试剂均为分析纯。
主要仪器有:KDN-102C型半自动定氮仪(上海纤检仪器有限公司);Allegra64R centrifuge型离心机(德国BECKMAN公司);DK-98-ⅡA型恒温磁力水浴锅(天津市泰斯特仪器有限公司);F-4500型荧光分光光度计(日本日立公司);DELTA320型pH计(梅特勒-托利多仪器有限公司);UV-240IPC型紫外可见分光光度计(日本岛津公司);DS-1型高速组织捣碎机(上海精科实业有限公司);JEM-1200EX型透射电子显微镜(日本日立公司)。
1.2 实验方法
1.2.1样品的制备
乳清浓缩蛋白自发形成纤维(WPC自发):按照前期研究的制备方法[2-3],取4.00 g乳清浓缩蛋白WPC-80溶于去离子水中,用6 mol/L HCl调节溶液的pH值至2.0并定容至100 mL,16 000g、4℃离心20 min,取中间清液,利用凯氏定氮法测定蛋白含量,随即用pH值2.0去离子水(用6 mol/L HCl调节去离子水的pH值至2.0)稀释蛋白质量分数至2.0%,90℃水浴加热10 h,取样并4℃冰箱保存。自发形成纤维过程中每隔1 h取样(0、1、2、3、4、5、6、7、8、9、10 h)并4℃冰箱保存。2.0%乳清浓缩蛋白溶液在pH值2.0、90℃水浴加热10 h,样品为成熟纤维。
纤维诱导乳清浓缩蛋白形成纤维(纤维诱导WPC):上述制备的2.0%乳清浓缩蛋白溶液中加入成熟纤维(纤维与乳清浓缩蛋白混合质量比为1∶3,蛋白终质量分数为2.0%),90℃水浴加热(0、1、2、3、4、5、6、7、8、9、10 h),取样并4℃冰箱保存。
乳清浓缩蛋白自发形成空白样(WPC空白):上述制备的2.0%乳清浓缩蛋白溶液中加入pH值2.0去离子水(乳清浓缩蛋白与pH值2.0去离子水混合体积比为1∶3),90℃水浴加热(0、1、2、3、4、5、6、7、8、9、10 h),取样并4℃冰箱保存。
纤维诱导乳清浓缩蛋白空白样(纤维空白):上述制备的2.0%纤维溶液中加入pH值2.0去离子水(纤维与pH值2.0去离子水混合体积比为1∶3),90℃水浴加热(0、1、2、3、4、5、6、7、8、9、10 h),取样并4℃冰箱保存。
1.2.2Th T荧光分析
参照AKKERMANS等[16]的方法并加以改进,用含有0.2 mol/L的NaCl、0.01 mol/L的磷酸缓冲液(pH值7.0)将硫黄素T(Th T)稀释成质量浓度为800 mg/L的溶液。将溶液用0.2 μm的滤膜过滤,除去不溶的Th T颗粒,即得到Th T储备液。将储备液置于包有铝箔的棕色试剂瓶中,4℃冰箱保存。测定时将Th T储备液用含有0.2 mol/L的NaCl、0.01 mol/L的磷酸缓冲液(pH值7.0)稀释50倍,即得到Th T工作液;取1 mL样品加入10 mL的Th T工作液中混匀,然后于荧光分光光度计下比色,在激发波长为460 nm,发射波长为490 nm以及狭缝宽度分别为5 nm和10 nm的条件下,测定Th T结合物的荧光强度。
纤维诱导过程中纤维自身Th T荧光强度也会变化,因此,纤维诱导过程中将去除纤维自身的影响。
纤维诱导乳清浓缩蛋白形成纤维过程中乳清浓缩蛋白的Th T荧光强度计算公式为
T=T1-T2
(1)
式中T1——纤维诱导WPC过程中的Th T荧光强度
T2——纤维热处理过程中自身的Th T荧光强度
为了保证蛋白终浓度一致,乳清浓缩蛋白自发形成纤维过程中Th T荧光强度计算公式为
T=T3-T4
(2)
式中T3——WPC自发形成纤维过程中的Th T荧光强度
T4——WPC热处理过程中自身的Th T荧光强度
1.2.3蛋白聚合率
取20 mL加热不同时间(0、1、2、3、5、8、10 h)蛋白质量分数为2.0%的样品溶液移入45 mL的离心管中,然后在16 000g离心20 min(4℃),弃去上清液,取离心管底部沉淀,利用凯氏定氮方法测定不同热处理时间的蛋白质质量浓度。蛋白聚合率计算公式为
C=Ct/C0×100%
(3)
式中C——蛋白聚合率,%
C0——0 h时样品蛋白质质量浓度,mg/mL
Ct——t时刻样品蛋白质质量浓度,mg/mL
纤维诱导乳清浓缩蛋白形成纤维过程中乳清浓缩蛋白的蛋白聚合率计算公式为
C=C1-C2
(4)
式中C1——纤维诱导WPC过程中的蛋白聚合率
C2——纤维热处理过程中自身的蛋白聚合率
为了保证蛋白终浓度一致,乳清浓缩蛋白自发形成纤维过程中蛋白聚合率计算公式为
C=C3-C4
(5)
式中C3——WPC自发形成纤维过程中的蛋白聚合率
C4——WPC热处理过程中自身的蛋白聚合率
1.2.4表面疏水性
参照相关ANS(8-苯氨基-1-奈磺酸)荧光探针法测定[17],并加以改进。用0.01 mol/L的磷酸缓冲液将不同热处理时间得到的蛋白溶液质量分数分别稀释为0.2%、0.1%、0.05%和0.025%。取已稀释后的蛋白溶液6 mL,加入20 μL的8 mmol/L ANS溶液,漩涡搅拌均匀,在室温(20℃)下避光15 min,在激发波长390 nm、发射波长470 nm和狭缝5 nm的条件下利用荧光分光光度计比色,测定荧光强度,以荧光强度为纵坐标,蛋白质溶液质量浓度为横坐标作图,初始斜率即为样品表面疏水性值。
纤维诱导乳清浓缩蛋白形成纤维过程中乳清浓缩蛋白的表面疏水性计算公式为
B=B1-B2
(6)
式中B1——纤维诱导WPC过程中的表面疏水性
B2——纤维热处理过程中自身的表面疏水性
为了保证蛋白终浓度一致,乳清浓缩蛋白自发形成纤维过程中表面疏水性计算公式为
B=B3-B4
(7)
式中B3——WPC自发形成纤维过程中的表面疏水性
B4——WPC热处理过程中自身的表面疏水性
1.2.5游离巯基
参照BEVARIDG等[18]的方法并加以改善。取400 μL不同热处理后的样品(20 mg/mL),加入至5 mL的Tris-Gly缓冲溶液(0.086 mol/L Tris,0.09 mol/L甘氨酸,0.004 mol/L乙二胺四乙酸,pH值8.0和8 mol/L尿素)中,然后向其中加入20 μL 5,5′-二硫代双(2-硝基苯甲酸) (DTNB)试剂,振荡混匀,室温下静止15 min,利用紫外分光光度计在412 nm波长下测定吸光度,以不加DTNB的溶液做空白调零。游离巯基浓度计算公式为
(8)
式中S——游离巯基浓度,μmoL/L
A412——在412 nm下的吸光度,计算时可用平均值
C′——固形物质量浓度,mg/mL
D——稀释系数
纤维诱导乳清浓缩蛋白形成纤维过程中,乳清浓缩蛋白的游离巯基浓度计算公式为
S=S1-S2
(9)
式中S1——纤维诱导WPC过程中的游离巯基浓度
S2——纤维热处理过程中自身的游离巯基浓度
为了保证蛋白终浓度一致,乳清浓缩蛋白自发形成纤维过程中游离巯基浓度计算公式为
S=S3-S4
(10)
式中S3——WPC自发形成纤维过程中的游离巯基浓度
S4——WPC热处理过程中自身的游离巯基浓度
1.2.6起泡性
参照STIEGER等[19]的方法测定蛋白质溶液的起泡能力和起泡稳定性并加以改进。样品用0.01 mol/L、pH值7.0的磷酸缓冲液稀释至0.15%,室温下用组织捣碎机10 000 r/min均质1 min,立即测定搅打后样品的体积,再测定放置30 min后样品的体积,通过相对溢出量和静止后稳定的泡沫体积比评价样品的起泡能力和泡沫稳定性,具体计算方法为
E=V0/Vi×100%
(11)
F=Vt/V0×100%
(12)
式中E——相对溢出量,%
F——泡沫稳定性,%
V0——起泡0 h时的泡沫体积
Vt——起泡t时间后的泡沫体积
Vi——起泡前最初液体的体积
纤维诱导乳清浓缩蛋白形成纤维过程中乳清浓缩蛋白的相对溢出量、泡沫稳定性计算公式为
E=E1-E2
(13)
F=F1-F2
(14)
式中E1、F1——纤维诱导WPC过程中的相对溢出量、泡沫稳定性
E2、F2——纤维热处理过程中自身的相对溢出量、泡沫稳定性
为了保证蛋白终浓度一致,乳清浓缩蛋白自发形成纤维过程中相对溢出量、泡沫稳定性计算公式为
E=E3-E4
(15)
F=F3-F4
(16)
式中E3、F3——WPC自发形成纤维过程中的相对溢出量、泡沫稳定性
E4、F4——WPC热处理过程中自身的相对溢出量、泡沫稳定性
1.2.7透射电镜
参照KREBS等[20]的方法,使用透射电子显微镜(TEM)测定样品的微观结构。将蛋白溶液质量浓度用去离子水稀释为1 mg/mL,将稀释液滴于透射电镜专用铜网上吸附20 min,然后把铜网移入滤纸上并干燥30 min,采用100 kV电压下用透射电镜进行分析。
1.2.8统计分析
试验数据采用Origin 8.6进行作图和SPSS 16.0软件对试验数据进行ANOVA方差分析,检验差异显著性(P<0.05)。数据均以平均值±标准差表示(n=3)。
2 结果与讨论
2.1 起泡性
2.1.1相对溢出量
本课题组前期研究已得出WPC自发形成纤维聚合物的起泡能力和起泡稳定性是其常规聚合物的2.40倍和2.00倍[13]。OBOROCEANU等[21]研究也得出同样的结果,WPI自发形成纤维状聚合物的起泡性和泡沫稳定性优于WPI常规聚合物。由图1可知,纤维诱导的WPC起泡能力远高于其自发形成纤维的起泡能力。纤维诱导乳清浓缩蛋白1 h和2 h的起泡能力分别是乳清浓缩蛋白自发形成纤维的1.36倍和1.41倍。热处理10 h,纤维诱导WPC起泡能力增量较WPC自发形成提高了32.35%。其次,在诱导过程中,纤维可以快速提高WPC的起泡能力,尤其在诱导前期(0~2 h)。纤维诱导WPC 2 h起泡能力已经略高于WPC自发形成纤维(10 h)的起泡能力,其变化量占整个诱导过程增量的80%,是WPC自发热处理2 h增量的6倍。结果说明纤维诱导的WPC起泡能力强于WPC自发形成纤维的起泡能力,并且可短时快速提高WPC的起泡能力。

图1 WPC与混入纤维的WPC热处理过程中 起泡能力的变化 Fig.1 Changes of foaming capacity of WPC and WPC mixed fibrils at different heating times
2.1.2泡沫稳定性
由图2可知,纤维诱导的WPC泡沫稳定性较WPC自发形成纤维的泡沫稳定性略有提高。但在诱导过程中,纤维仍具有快速提高WPC泡沫稳定性的能力(诱导前期0~2 h);纤维诱导WPC 2 h的泡沫稳定性就已高于WPC自发形成纤维(10 h)的泡沫稳定性,其变化量占整个诱导过程增量的79.24%,是WPC自发热处理2 h增量的6.11倍。结果说明混入成熟纤维可快速提高WPC的泡沫稳定性。纤维诱导的WPC起泡性高于非诱导情况下WPC自发形成的起泡性,可能是因为诱导与自发两种处理手段导致WPC自组装聚合形成的纤维能力不同。

图2 WPC与混入纤维的WPC热处理过程中泡沫 稳定性的变化 Fig.2 Changes of foaming stability of WPC and WPC mixed fibrils at different heating times
2.2 Th T荧光分析
自发和纤维诱导的WPC在pH值2.0、90℃热处理条件下,逐渐聚合形成纤维;此过程中β-折叠数量不断增加,硫磺素T(Th T)是一种能与叠加的β-折叠特异性结合的染料,结合后的荧光强度在一定范围内随着β-折叠数量的增加而增强[22],因此,通常用此方法间接反映纤维形成情况。纤维诱导的WPC在热处理过程中荧光强度明显高于WPC自发形成(图3a)。在诱导过程中,纤维诱导的WPC荧光强度增量略高于WPC自发形成,也有文献得出相同结果[14,23]。但二者纤维形成速率存在差异(图3b,图中不同字母表示组内有显著性差异(P<0.05))。WPC自发形成纤维过程中荧光强度变化量主要集中在2~5 h内;而纤维诱导的WPC荧光强度变化量主要集中在诱导前2 h。纤维诱导WPC 2 h的荧光强度已接近WPC自发热处理10 h的荧光强度,其变化量占整个诱导过程增量的80.56%,是WPC自发热处理2 h增量的12.04倍。由此结果可知较WPC自发形成纤维,成熟纤维的混入可快速诱导WPC形成纤维,缩短纤维形成时间,改善WPC起泡性。

图3 WPC与混入纤维的WPC热处理过程中Th T荧光强度的变化 Fig.3 Changes of Th T fluorescence intensity of WPC and WPC mixed fibrils at different heating times
2.3 透射电镜
纤维诱导WPC与WPC自发形成纤维过程中其微观形态存在差异,微观形态差异可反映聚合能力的强弱(图4)。WPC自发形成纤维过程中,球形蛋白颗粒不断聚合形成低聚物,最终在热处理10 h时形成细长的、有分支的成熟纤维(图4c)。而纤维诱导2 h时就已形成纤维(无低聚物存在),纤维形成时间由10 h缩短至2 h。在pH值2.0、90℃热处理条件下,纤维诱导WPC形成纤维的微观形态与WPC自发形成纤维的微观形态无明显差异,均形成有分支的棒状结构,但纤维可以加速诱导WPC形成纤维,缩短纤维形成时间。

图4 WPC与混入纤维的WPC热处理过程中形成聚合物的透射电镜图 Fig.4 Transmission electron micrographs of WPC and WPC mixed fibrils
2.4 蛋白聚合率
为了定量比较纤维诱导和自发形成纤维过程中形成的WPC纤维量,研究了混入纤维的WPC在热处理过程中蛋白聚合率(图5)。由图可知,纤维诱导的WPC在热处理过程中蛋白聚合率明显高于WPC自发形成。热处理10 h,纤维诱导的WPC蛋白聚合率变化量较WPC自发形成提高了33.79%。同时在诱导过程中,纤维诱导WPC 2 h的蛋白聚合率接近WPC自发热处理10 h的蛋白聚合率,其变化量占整个诱导过程变化量的69.10%,是WPC自发热处理2 h变化量的3.59倍。由此可知较WPC自发成纤维,混入成熟纤维可以快速诱导WPC形成纤维,并且提高纤维量,从而提高了WPC的起泡性。
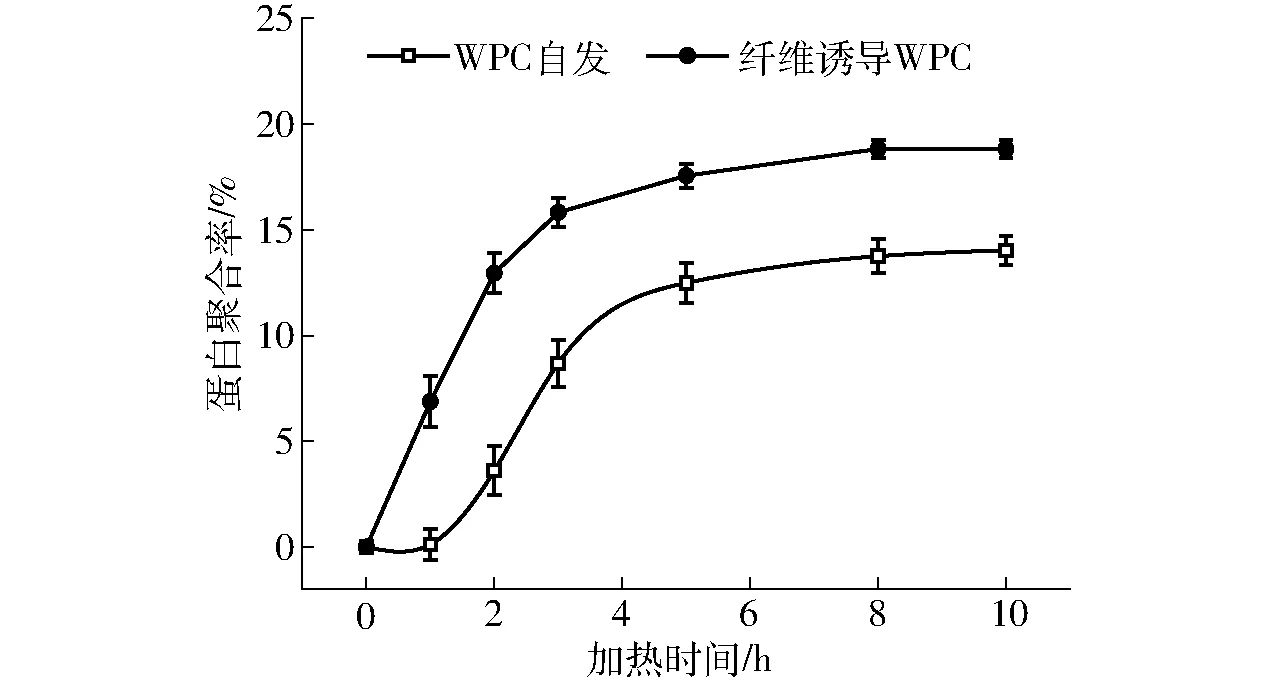
图5 WPC与混入纤维的WPC热处理过程中 蛋白聚合率的变化 Fig.5 Changes of protein polymerization rate of WPC and WPC mixed fibrils
2.5 聚合驱动力
蛋白质在一定条件下加热通过某些驱动力而发生聚合,这些驱动力包括共价作用和非共价作用。蛋白质在常规pH值条件下加热主要通过共价作用(巯基-二硫键的交换作用)相互聚合,而在酸性pH值(远离等电点)条件下加热,蛋白质主要通过疏水作用、氢键、范德华力等非共价作用聚合形成纤维状聚合物。WPC自发和纤维诱导的WPC聚合过程中主要驱动力的差异,主要是表面疏水性和游离巯基浓度的变化。纤维诱导的WPC与WPC自发形成纤维过程中聚合驱动力的变化趋势一致,表面疏水性均呈现先上升后下降的趋势(图6a)。但纤维诱导过程中表面疏水性变化主要集中在诱导0~1 h内,其变化量占整个诱导过程增量的74.78%,是WPC自发形成变化量的1.32倍。纤维诱导的WPC与WPC自发形成纤维过程中游离巯基含量均缓慢降低[24](图6b)。但纤维诱导的WPC游离巯基变化量同样在诱导0~1 h内最大,其变化量占整个诱导过程变化量的36.93%,是WPC自发形成变化量的2.86倍。从以上结果可知,纤维诱导WPC形成纤维与WPC自发形成纤维过程中,表面疏水性是主要驱动力,二硫键作用很小[3,25]。同时,较WPC自发形成纤维,纤维诱导过程中的聚合驱动力可在短时内(0~2 h)快速发生变化,加速纤维的形成。

图6 WPC与混入纤维的WPC热处理过程中聚合驱动力的变化 Fig.6 Changes of driving aggregation force of WPC and WPC mixed fibrils
3 结论
(1)与乳清浓缩蛋白自发形成纤维相比,纤维诱导的乳清浓缩蛋白在热处理过程中可显著提高乳清浓缩蛋白的起泡性。
(2)纤维还具有短时快速提高乳清浓缩蛋白起泡性的能力,尤其在诱导前期(0~2 h)。这是因为诱导与自发两种处理手段导致乳清浓缩蛋白自组装聚合形成的纤维能力不同所致。纤维可快速诱导乳清浓缩蛋白形成纤维,缩短纤维形成时间并提高纤维量,从而改善乳清浓缩蛋白的起泡性。
