纳米碳酸钙粒径测试的研究
2018-08-13莫英桂韦健毅满治成黎宇平
莫英桂,朱 勇,韦健毅,满治成,黎宇平
(广西华纳新材料科技有限公司,广西南宁530103)
纳米碳酸钙是一种无机超细粉末材料,具有粒径小、比表面积大等特点,可应用于诸多领域[1-4]。不同的应用对纳米碳酸钙的物化性质要求各异,纳米碳酸钙的白度、形貌、比表面积和粒径等直接影响填充产品性能[5]。在大多数情况下,纳米碳酸钙的粒径起着至关重要的作用。
由于纳米碳酸钙颗粒小、比表面积大、表面能高、易发生团聚,因此在粒径测试时需区分一次粒径和二次粒径(团聚体粒径)。在纳米碳酸钙生产和应用中,比表面积是十分重要的实用化质量指标,其大小与晶体粒径密切相关,可间接反映纳米碳酸钙颗粒的大小。比表面积越大,粒径越小,反之亦然。
笔者通过电子显微镜法、X射线衍射法、氮气吸附BET法、压汞法和激光散射法表征了纳米碳酸钙颗粒大小,比较并讨论了纳米碳酸钙粒径测试方法的优缺点。
1 实验部分
1.1 原料、试剂与仪器
原料与试剂:自制纳米碳酸钙、皂粒8020(上海制皂集团有限公司)、无水乙醇(天津市致远化学试剂有限公司,分析纯)
仪器:TG 209 F3 Tarsus型热重分析仪、H-7500型透射电子显微镜(TEM)、SUPRA Sapphire型扫描电子显微镜(SEM)、Smart Lab型X射线衍射仪(XRD)、GeminiⅦ 2390 型比表面积分析仪(BET)、AutoPore IV 9500 型压汞仪(MIP)、Mastersizer 3000E型激光粒度分析仪。
1.2 实验方法
1.2.1 纳米碳酸钙制备方法
采用碳化法制备纳米碳酸钙浆液。将一定量的皂粒加热溶解后,加入碳酸钙浆液中,50~60℃下乳化0.5 h,脱水、烘干至水质量分数低于0.6%,粉碎后得到纳米碳酸钙。
1.2.2 纳米碳酸钙表面处理剂实际包覆量测试方法
利用热重法根据纳米碳酸钙温度-质量变化关系来测定其表面处理剂的实际包覆量。测试条件:空气气氛,测试温度为500℃,升温速率为10℃/min。
1.2.3 电子显微镜分析方法
透射电子显微镜:取适量试样,置于烧杯中,以无水乙醇作分散剂,制成质量分数为10%的纳米碳酸钙分散液。将烧杯置于超声波振荡仪中,超声分散5 min后,取1~2滴分散液于制样铜网(孔径为0.011 4 μm)自然干燥后进行TEM表征。加速电压为 120 kV,放大倍数为 10 000~50 000。
扫描电子显微镜:将少量粉末试样均匀地撒在样品座的导电胶上,再用洗耳球将未黏住或黏结不牢固的粉末吹去,对样品做喷金处理后进行SEM表征。加速电压为2 kV,放大倍数为10 000~50 000。
使用图像处理软件Image J 1.47V分析图像,获得纳米碳酸钙颗粒的大小分布。
1.2.4 X射线衍射分析方法
将样品按X射线衍射仪粉末制样要求制样后进行测试,衍射条件:Cu靶Kα辐射,管电压为40kV,管电流为40 mA,扫描步长为0.02°,扫描速度为2(°)/min,扫描角度范围为 2θ=10~70°。使用 Scherrer公式计算晶粒尺寸。
1.2.5 比表面积测试方法
比表面积测试采用氮气吸附BET法。称量约0.100 g样品放入样品管(样品管质量为m1),150℃下干燥1 h,冷却后称量质量记为m2,并计算实际粉体样品质量m=m2-m1。将样品管安装到进气口。打开测试软件,输入实际粉体质量及试样名称后测试。测试结束后记录BET比表面积数据。测试条件:N2吸附,吸附温度为-196.15℃。
1.2.6 压汞仪测试方法
测试前将纳米碳酸钙置于110℃烘箱中干燥1 h。称量0.5 g烘干后的样品,装入粉末膨胀计,将膨胀计装入低压站进行低压分析。低压分析结束后,将膨胀计取出称量质量,再将膨胀计装入高压站,进行高压分析。测试压力范围为15~228 MPa。
1.2.7 激光粒度仪测试方法
在循环分散烧杯中倒入600 mL无水乙醇,选择分散剂并输入光学参数,初始化仪器及调节测量背景后,加入测试样品,使样品遮光强度为10%~15%,并开启超声分散(1 min),待遮光强度稳定后测量纳米碳酸钙粒径。
2 结果与讨论
本文基于不同的纳米碳酸钙进行粒径表征并讨论,纳米碳酸钙样品具体信息见表1。其中,NCC-01~NCC-06为同样的纳米碳酸钙,表面处理剂用量不同;NCC-07~NCC-08为粒径不同的纳米碳酸钙,表面处理剂用量与NCC-03相同。
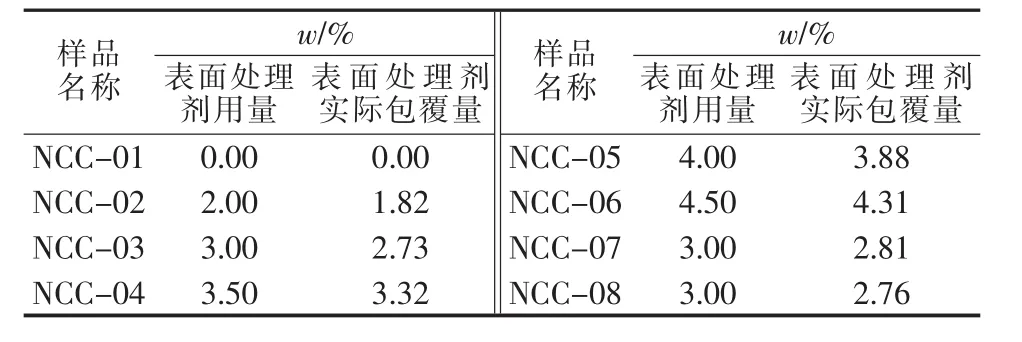
表1 纳米碳酸钙样品相关数据
2.1 电子显微镜法
图1是纳米碳酸钙的TEM照片。由图1可见,纳米碳酸钙颗粒重叠严重,颗粒边界模糊,使用图像处理软件分析粒径时不易确定颗粒边界,难以确定晶粒尺寸 (一次颗粒)和团聚体尺寸 (二次颗粒)。TEM制样时使用无水乙醇分散纳米碳酸钙,将悬浮液滴至铜网,纳米碳酸钙晶粒随乙醇挥发而固定于铜网。造成颗粒重叠的可能原因:1)纳米碳酸钙本身分散性较差;2)无水乙醇可能溶解了部分纳米碳酸钙表面处理剂,乙醇挥发过程中形成的晶粒间液桥可能导致纳米碳酸钙再次团聚。TEM观察到的团聚体难以确认是制样过程产生的,还是纳米碳酸钙样品本身的原始团聚体。此外,TEM放大倍数升高会限制视野,照片上可见粒子数减少,很难获得足够多的单分散颗粒进行粒径统计分析[6-7]。因此,TEM不适合用于统计分析表征纳米碳酸钙粒径,且可用于直接观察局部个别纳米碳酸钙粒子的大小和形貌。

图1 纳米碳酸钙的TEM照片
图2是纳米碳酸钙样品的SEM照片。由图2可见,样品图像清晰,颗粒边界易于辨认。每个样品选取3个视野的电镜照片做图像处理分析,结果见表2。由表2可见,NCC-01~NCC-06的平均一次粒径很接近,为76 nm左右;NCC-07的一次粒径为90.4 nm;NCC-08的一次粒径为103.4 nm。3个表面处理剂用量相同的纳米碳酸钙比较,平均一次粒径大小依次为dSEM(NCC-08)、dSEM(NCC-07)、dSEM(NCC-03)。 扫描电子显微镜测定纳米碳酸钙的粒径,可对其进行统计分析,但表征结果存在取样代表性的问题。

图2 纳米碳酸钙的SEM照片
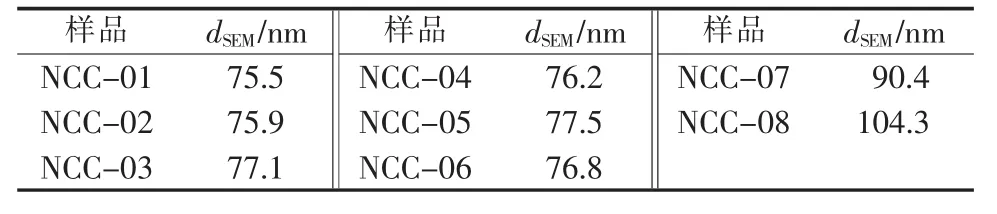
表2 扫描电镜粒径
2.2 X射线衍射粒径测试
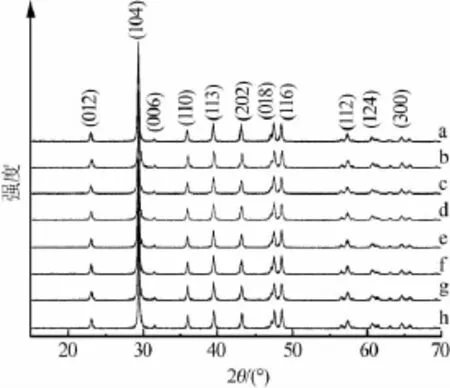
图3 纳米碳酸钙XRD谱图
图3是纳米碳酸钙样品的XRD谱图。由图3可见,样品的主要特征衍射峰位置基本重合,为方解石型碳酸钙(PDF 47-1743)。原始X射线衍射谱经去背底,线形光滑,双线分离后,用X射线衍射硅粉末标准物质对仪器宽化校正。根据Scherrer公式,由(104)晶面衍射峰可计算出纳米碳酸钙的晶粒尺寸dXRD:

式中,K 为 Scherrer常数,方形粒子取 0.943[8-9];λ 为入射线波长,0.154 06 nm;β为样品实际衍射峰半高宽,计算时需转化为弧度;θ为衍射角,°;dXRD为纳米碳酸钙平均粒径,nm。
采用式(1)计算纳米碳酸钙的平均粒径,结果见表3。将表3和表2对比可以发现,dXRD略小于dSEM。对比可知,XRD得到的平均粒径接近纳米碳酸钙的一次粒径,且表面处理剂用量不影响XRD测定结果。这一方法具有简单、快速的优点。

表3 XRD测试粒径
2.3 气体吸附法
实验采用氮气吸附BET法测试纳米碳酸钙的比表面积,并通过式(2)计算出纳米碳酸钙比表面积的当量粒径:

式中,ρ为碳酸钙密度,2.71 g/cm3;SSA为纳米碳酸钙的比表面积;K为形状因子,粒子为球形或者立方形时,K=6.0[10]。
表4为纳米碳酸钙的比表面积及按式(2)计算所得纳米碳酸钙比表面积的当量粒径。由表4可见,纳米碳酸钙的表面处理剂影响其比表面积,随表面处理剂用量增大,比表面积明显减小,计算得到的纳米碳酸钙比表面积当量粒径增大。同一粒径下未经表面处理的纳米碳酸钙比表面积最小,比表面积当量粒径最大。相同表面处理剂用量的条件下,NCC-03、NCC-07和NCC-08的比表面积当量粒径大小关系:dBET(NCC-08)>dBET(NCC-07)>dBET(NCC-03),与dSEM、dXRD关系一致。
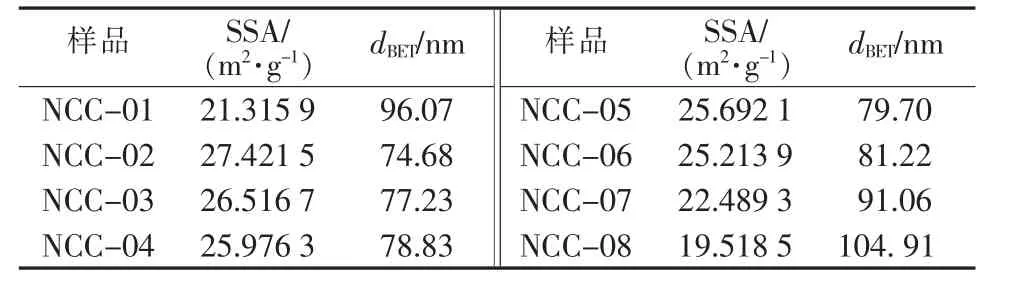
表4 BET法测试结果
NCC-02~NCC-06是对同样的纳米碳酸钙浆料表面处理得到的,其原始表面形态及孔径分布情况相同。纳米碳酸钙颗粒粒径小,表面能高,处于不稳定状态,易形成由多个颗粒组成的较大的团聚体。经表面处理的纳米碳酸钙表面形成了有机分子膜,阻碍了碳酸钙颗粒之间直接接触,并降低表面张力,可减少团聚发生。此外,纳米碳酸钙表面孔隙被有机分子修饰,部分内孔可能在表面处理过程中被表面处理剂填充。一般来说,表面处理剂用量越大,则对纳米碳酸钙包覆越完整,对其表面的孔隙修饰作用越大。因此,经表面处理的纳米碳酸钙的比表面积随表面处理剂用量增大有明显减小的趋势。而未经表面处理的纳米碳酸钙,颗粒间直接接触,在干燥过程中形成晶桥,并使得部分纳米碳酸钙颗粒结块成粒径较大的团聚体,表面积减小。
与dSEM和dXRD相比,经表面处理的纳米碳酸钙dBET略大于前两者,而未经表面处理的纳米碳酸钙粒径则大很多。可见BET法是一种有效表征纳米碳酸钙一次粒径的方法,但颗粒团聚也会给测试带来误差。
2.4 压汞法
压汞法是目前测试粉体比表面积和孔径分布基本的方法,也是应用广泛的方法之一。对纳米碳酸钙进行压汞测试,结果见表5。由表5可见,对同样的纳米碳酸钙浆料表面处理得到的NCC-02~NCC-06,总孔容随表面处理剂用量的增大而减小,这也佐证了表面处理剂用量与其对纳米碳酸钙表面孔隙的修饰作用的正相关关系;比表面积随表面处理剂用量的增大而增大。而未经表面处理的NCC-01总孔容最大,比表面积最小。表面处理剂用量相同(3%)的其比表面积由大到小顺序:NCC-03、NCC-07、NCC-08,与BET法比表面积关系一致,但绝对数值相差较大,总孔容则随比表面积减小而减小。

表5 MIP测试结果
图4为进汞-退汞曲线。图5为压汞微分曲线。由图4可知,未经表面处理的纳米碳酸钙(NCC-01)大孔较多,所以导致其总孔容最大,而比表面积最小。由图5可知,NCC-01大孔多,中微孔少;而NCC-02~06大孔少,中微孔多。在压汞测试过程中的高压下,中微孔会变形甚至崩塌,导致测试结果误差较大。与BET法相比,压汞法测试纳米碳酸钙的比表面积误差较大。

图4 进汞-退汞曲线

图5 压汞微分曲线
2.5 激光散射法
纳米碳酸钙团聚体在一般条件下不易解聚,在填充体系中可能以二次颗粒的形式存在,二次颗粒大小影响其应用性能[11]。激光散射法是表征纳米材料二次颗粒粒度的一种有效方法[11]。表6为不同纳米碳酸钙的激光散射粒径参数。由表6可见,残差值均小于3%,表明测试光学参数正常,测试模型准确,粒径分布曲线见图6。由图6可见,纳米碳酸钙的二次颗粒为微米级,测试结果与文献[12]的实验结果接近。经表面处理的纳米碳酸钙粒度受表面处理剂用量影响[11],其平均粒径(D50)均大于未表面改性处理的纳米碳酸钙的平均粒径,但未见激光散射法测试纳米碳酸钙的粒径结果与表面处理剂实际包覆量呈现明显规律。

表6 纳米碳酸钙二次粒径参数
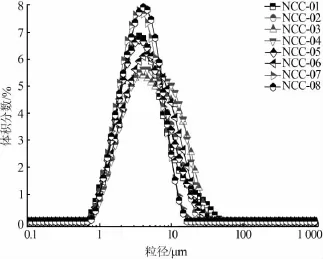
图6 激光散射法粒径分布
为验证激光散射法测试结果的有效性和准确性,选用NCC-07做重复测试,结果见表7。由表7可见,2个样品的D10、D50和D90相对标准偏差均小于有效测量的偏差,说明测试结果准确度较高。
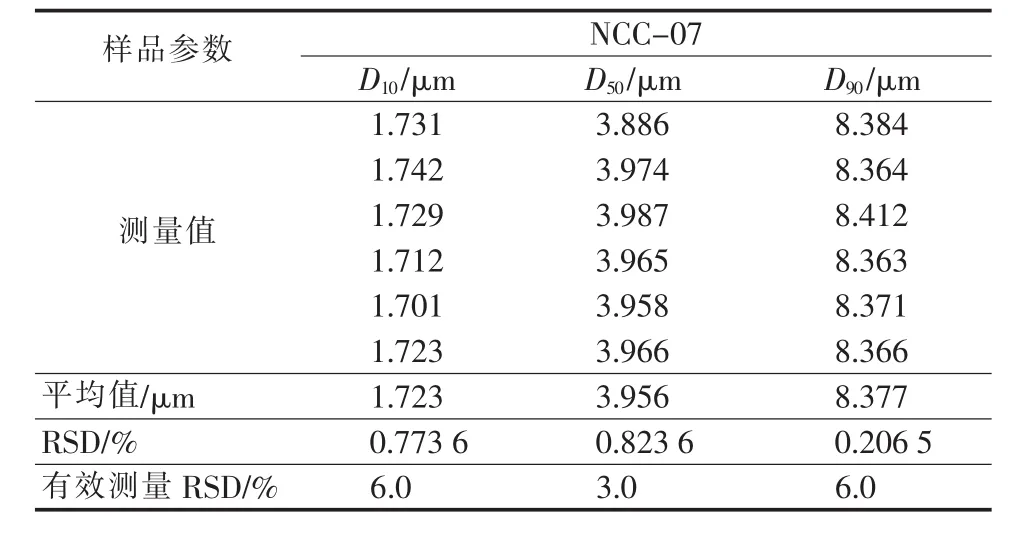
表7 重复实验测试结果
3 结论
1)SEM、XRD法和BET法测试得到的纳米碳酸钙粒径结果相近,SEM法测试纳米碳酸钙粒径的同时可观察其粒子形貌,但该方法存在取样代表性问题;BET法测试结果受纳米碳酸钙表面处理影响。2)TEM可用于直观观察纳米碳酸钙的形貌和粒径,不适用于图像统计分析的粒径分析。3)压汞法测试纳米碳酸钙比表面积结果误差较大;4)激光散射法可有效表征纳米碳酸钙的二次粒径。
