非贵金属单原子催化剂的研究进展
2018-08-10吴耿煌荣峻峰达志坚
吴耿煌, 黄 凰, 荣峻峰, 达志坚
(1.中国石化 石油化工科学研究院, 北京 100083; 2.国民核生化灾害防护国家重点实验室, 北京 102205)
催化剂作为催化技术的核心,能够有效地调控化学反应的路径,在更为温和的条件下,高选择性地获得目标产品[1]。工业催化中催化剂与反应物通常处于不同的相态,因此催化过程主要涉及多相催化。其中,负载型金属催化剂由于具有较高的催化活性与选择性,被广泛应用于重要的化工产品的生产过程。对于负载型金属催化剂,金属颗粒的尺寸是影响其催化性能最重要的因素之一[2,3]。表面原子与总原子数之比随着粒径的变小而急剧增大,产生了更多的配位不饱和金属中心。此外,当颗粒尺寸下降到一定值时,费米能级附近的电子能级由准连续能级变为分离能级,产生量子尺寸效应;同时粒径的减小还增强颗粒与载体之间的相互作用,这都直接影响到催化剂的性能[4-6]。直观上看,为了最大限度地利用金属材料,将金属以单原子的形式分散在载体表面并且作为活性位点,是多相催化所能达到的极限[7-9]。
2011年,张涛院士团队利用氧化铁与铂原子之间的强相互作用,成功制备出首例具有高活性和高稳定性的单原子铂催化剂Pt1/FeOx[10],引发了国际上对单原子催化剂的研究热潮。单原子催化剂是指催化剂中活性组分完全以孤立的单个原子的形式存在,并通过与载体作用或与第二种金属形成合金得以稳定。相比于纳米/亚纳米催化剂,单原子催化剂具有诸多优势:(1)最大限度地提高金属原子利用率;(2)活性位点的组成和结构单一,可避免因活性组分组成和结构不均匀导致的副反应;(3)兼具均相催化剂均匀单一的活性中心和多相催化剂结构稳定易分离的特点,因此单原子催化剂有望成为连接均相催化与非均相催化的桥梁[11,12]。继Pt1/FeOx的成功制备之后,高性能的Ir/FeOx[13],Au/CeO2[14]和Pd/ZnO[15]等贵金属单原子催化剂的研究也取得了突破。然而考虑到贵金属的稀缺性,发展可能替代贵金属催化剂的高性能非贵金属单原子催化剂显然具有更大的价值空间。在自然界,固氮酶中的Mo,血红素中的Fe,叶绿素中的Mg等都可以看成是高效的非贵金属单原子催化剂[16]。最新的研究还表明,基于Fe、Co等的非贵金属单原子催化剂在某些催化反应中所体现的性能足以媲美甚至优于贵金属催化剂[17,18]。鉴于非贵金属单原子催化的研究近年来取得诸多进展及巨大的发展潜力,在本文中,笔者重点介绍了非贵金属单原子催化剂的制备、表征方法及其在电催化、加氢及脱氢反应、液相氧化反应、光催化还原CO2等领域的应用研究进展,同时展望了非金属单原子催化剂的发展趋势。
1 非贵金属单原子催化剂的制备方法
1.1 原子层沉积法
原子层沉积(ALD)也称为原子层外延,是一种将气相前驱体脉冲交替地通入反应腔并在基体表面发生化学吸附及化学反应,形成单原子沉积膜的方法。ALD具有沉积参数高度可控、优异的沉积均匀性和一致性等优点,被广泛应用于纳米材料的制备[19]。Li等[20]以热稳定性良好的Zr基金属有机框架(MOF)NU-1000为沉积载体,双(N,N′-二-叔-丁基乙脒基)镍为前驱体,在NU-1000的Zr6位点上均匀沉积了原子级别分散的单位点Ni催化剂。其中Ni通过O与Zr键合而稳定,Ni的化学态接近二价的离子态。随后他们又以ALD法及水热沉积法分别在 NU-1000 上沉积了单位点的Co[21]。同单位点的Ni一样,这两类Co基催化剂中Co的价态均为二价离子态。此外,Qiu等[22]通过化学气相沉积(CVD),在纳米多孔Ni载体表面沉积多孔石墨烯后用HCl将Ni载体移除,残留了少量的Ni与C直接键合,以单原子的形式嵌入石墨烯表面。Zhang等[23]通过水热沉积,将Co以单位点的形式稳定在MOF-525(Zr)的卟啉环空穴中,制备了单位点MOF-525-Co催化剂。总体而言,目前各类沉积法通常需预先制备特定有序结构的载体单元,合成步骤较为繁琐且使用的有机配体价格较为昂贵,不适用于大批量制备非贵金属单原子催化剂。
1.2 电弧放电法
电弧放电法是一种制备碳基金属复合材料的重要方法[24]。Zhang等[25]采用改进的电弧放电法成功制备了石墨化碳层负载单原子Nb的催化剂。图1[25]为电弧放电法制备石墨化碳层负载单原子Nb催化剂的装置示意图,Nb棒为阳极,C棒为阴极,CH4为气相C源,工作电压30 V,工作电流90 A。需要指出的是,所制备的碳基催化剂还负载了一定量的碳包覆NbC颗粒。由于电弧放电法制备的催化剂容易产生包括纯碳材料、碳包覆金属纳米颗粒等在内的杂质且不易分离,因此这类方法并不适合批量制备高纯度的碳载单原子催化剂。

图1 电弧放电法制备石墨化碳层负载单原子 Nb(Nb-in-C)的装置示意图[25]Fig.1 Schematic diagram of the synthesis equipment used to produce Nb-in-C complex[25]
1.3 高能球磨法
高能球磨法利用机械能来诱发化学反应或诱导材料组织、结构和性能的变化,以此来制备新材料[26]。Deng等[27]在Ar气氛保护的条件下,高能球磨酞菁铁分子与石墨烯纳米片(Graphene nanosheets, GN),利用N与GN的C形成强的共价键,使得N作为“锚”来稳定配位不饱和的Fe中心。通过改变酞菁铁与GN的比例,制备了Fe负载量(质量分数)分别为1.5%、2.7%以及4.0%的3种单原子催化剂。进一步利用高分辨透射电镜(HRTEM)、高角度环形暗场扫描透射电镜(HAADF-STEM)以及低温扫描隧道显微镜等表征手段证实了Fe通过与4个N配位形成FeN4中心,并限域在石墨烯骨架中。此后他们还分别以酞菁锰、钴、镍、铜为金属前驱体,成功制备了单位点的MnN4/GN、CoN4/GN、NiN4/GN、CuN4/GN,证实了以酞菁配位的非贵金属盐与GN为前驱体通过球磨法制备对应的N配位的非金属单位点催化剂具有较好的普适性[28]。
1.4 液相化学还原法
液相化学还原法是指利用还原剂的还原性,在液相中将金属阳离子还原为对应的金属单质的方法。通过改变溶剂、还原剂、金属前驱体、表面活性剂、载体以及反应温度与时间,可以十分有效地调控金属纳米材料的组成、形貌与尺寸[29]。Long等[30]以TiO2纳米片为载体,维生素C为还原剂,水相中还原Pd、Cu金属盐,将具有不同组成比例的PdxCu1合金纳米颗粒负载于TiO2表面(PdxCu1-TiO2)。X射线吸收精细结构(XAFS)分析表明,当Pd/Cu摩尔比大于7时,合金中的Cu以单原子的形式与Pd成键,Cu之间无金属键作用,并且由于Pd的保护作用,Cu以金属态的形式稳定。尽管利用液相化学还原法通过调节金属前驱体的比例以制备非贵金属单原子催化剂的方法相对较为简单,但此方法的普适性还有待进一步的验证。
1.5 沉积-沉淀法
沉积-沉淀法是指将需负载的金属盐溶液与载体在搅拌条件下形成均匀的悬浮液,控制一定的温度和pH值,使金属沉积在载体表面,随后进行过滤、洗涤、干燥、焙烧等处理,得到负载金属的催化剂[31]。SiO2是最常用一类载体,其表面丰富的含O官能团可以与金属发生键合,有效地稳定金属组分[32]。Zhu等[33]采用尿素辅助的沉积-沉淀法制备了SiO2负载的单原子Cu催化剂(Cu/SiO2-UH),Cu的负载量高达15%(质量分数)。尿素在制备过程中起到2个作用,一是加热过程中发生水解,提高溶液pH值,促进了硅醇基的去质子化;二是水解产生的NH3与Cu2+配位形成铜氨离子。铜氨离子进一步与去质子化的硅醇基发生键合,形成了Cu-O-Si共价键,实现了Cu的单原子分散。随后用H2对催化剂进行还原处理,还原后Cu主要为Cu0以及Cu+2种价态,且HRTEM证实Cu仍为单原子分散状态。此外将可溶性铜盐替换成锰盐、钴盐、镍盐和锌盐,也可制备出对应的SiO2负载的非贵金属单原子催化剂。
1.6 原位活化法
原位活化法与前面所述方法的区别在于单原子催化剂是由新鲜催化剂(非单原子催化剂)在反应过程中原位活化而制得。Guo等[34]将SiO2与Fe2SiO4球磨混合后进一步高温熔化,用HNO3沥滤,干燥获得了Fe©SiO2催化剂并将该催化剂应用于甲烷制乙烯、芳烃的反应中。研究发现,在1090℃的高温下反应60 h 后,新鲜催化剂上的FeOx颗粒发生结构重构,Fe在反应过程中重新以单一位点的形式分布在SiO2基质上;XAFS进一步验证了反应过程中Fe镶嵌在Si 和C 之间形成了键合作用,保证该催化剂高温下的稳定。此外,Fan等[35]以Ni-MOF为前驱体,经过700℃高温碳化后,用HCl沥滤,干燥获得了石墨碳包覆Ni纳米颗粒核壳结构催化剂并将该催化剂应用于电催化析氢(HER)反应,通过HAADF-STEM直观地证实在电化学扫描过程中,催化剂发生原位活化,核层Ni纳米颗粒逐渐溶解,以单原子的形式重新嵌入石墨化碳层中。由于原位活化法存在一定的特殊性,因此利用此方法有目的地制备非贵金属单原子催化剂仍存在较大难度。
1.7 高温裂解法
Co和Fe通过与碳载体上的N配位所形成的单原子分散的Co-N-C以及Fe-N-C是两类非常重要的非贵金属单原子催化剂[18,36]。Co(Fe)-N-C催化剂通常采用高温裂解法制备,即将含N、C的有机配体配位的金属前驱体与碳载体或其他模板载体在600~900℃惰性气氛下高温裂解碳化,是目前制备非贵金属单原子催化剂最主要的方法。Liang等[37]以维生素B12(VB12)、聚苯胺铁络合物(PANI-Fe)为前驱体,SiO2纳米颗粒、有序介孔的SBA-15以及层状结构的蒙脱石3种SiO2材料为模板,经高温裂解碳化后用HF除去模板及金属纳米颗粒,得到Co-N-C及Fe-N-C催化剂。如图2[37]所示,通过选用不同结构的模板,可有效地调控Co(Fe)-N-C孔结构及比表面积,实现对催化剂性能的调控。Shen等[38]同样以SBA-15为模板,浸渍FeCl3并与CCl4、乙二胺、升华S水热混合后高温裂解,用HF去除模板,制备了S掺杂的Fe-N-C催化剂。XAFS表明,Fe与N的平均配位数为2,主要以FeN2位点的形式负载于碳载体上。除了SiO2材料外,Mg(OH)2及MgO也可以作为一类牺牲载体,用于Co(Fe)-N-C的制备。Liu等[39]将醋酸根、菲啰啉共同配位的Co(phen)2(OAc)2负载于Mg(OH)2后,700℃高温裂解碳化,用HNO3沥滤MgO,干燥后获得了质量分数高达3.6%的Co-N-C单原子催化剂。此类方法还可用于制备质量分数为1.8%的Fe-N-C单原子催化剂[40]。此外,Zhu等[41]以Te纳米线为模板,制备了碳纳米管(CNT)气凝胶负载的Fe-N-C催化剂。由于Te纳米线在高温下易挥发,因此模板在高温裂解的过程中自动移除,无需再经沥滤、干燥等后处理步骤。除了以非碳材料作为牺牲模板外,也可直接将炭黑[42]、石墨烯[43]、CNT[44,45]以及碳链聚合物[46]等碳载体负载有机配体配位的金属盐,经高温裂解、强酸沥滤以制备单原子的Co(Fe)-N-C催化剂。

图2 模板法制备介孔Co(Fe)-N-C催化剂的示意图[37]Fig.2 Schematic illustration of templating synthesis of mesoporous Co(Fe)-C-N catalysts[37]
沸石咪唑酯骨架结构材料(ZIF)是一种新型的多孔晶体材料,属于具有特定配体的MOF材料。ZIF中,有机咪唑酯通过N的配位作用,交联连接到过渡金属上,形成一种四面体框架[47]。由于其有序的骨架结构,既有N配位可调控的金属中心(Fe、Co、Zn等),且配体可作为碳源,因此由ZIF制备单原子Co(Fe)-N-C催化剂具有显著的优势与潜力。Zitolo等[48]以负载Fe(phen)2(OAc)2的ZIF-8为前驱体,经过1050℃高温裂解,用H2SO4沥滤、干燥获得了Fe质量分数为1.0%的Fe-N-C催化剂。Yin等[49]在ZIF的合成过程中同时加入Co、Zn盐,制备了双金属的CoZnZIF,如图3[49]所示,通过调控Zn与Co的比例,可有效调控ZIF中Co原子节点的距离;此外Zn在高温裂解过程中挥发,配位的前驱体转化为额外的N掺杂位点有利于进一步稳定Co原子,这两种作用有效避免Co在高温下的团聚。利用这种方法,成功制备了Co负载量>4%的单原子Co-N-C催化剂。相同的方法还可用于制备单原子Ni-N-C[50]和Fe-N-C催化剂[51]。与Co(Ni)Zn体系不同的是,FeZn体系ZIF合成的过程中Fe、Zn没有形成双金属ZIF,前驱体Fe(acac)3被封装在ZIF-8(Zn)的分子笼内。由于孔径的限制,每个分子笼只能捕获1个Fe(acac)3,这种特殊的结构有效避免了高温裂解过程中Fe的团聚。此外,Zhang等[52]则合成了粒径可控的双金属FeZnZIF,并经高温裂解后,无需酸洗沥滤,直接制备单原子Fe-N-C催化剂。

图3 氮掺杂碳负载的Co纳米颗粒(CoNPs-N/C)及 单原子Co-N-C的制备过程示意图[51]Fig.3 Schematic illustration of the formation of Co NPs-N/C and Co-N-C[51]
最近,Zhang等[53]还提出了一种基于金属(氢)氧化物@聚合物核壳结构制备金属单原子催化剂的方法。如图4所示,将FeOOH纳米棒表面包覆聚多巴胺(PDA)后,经高温碳化,HCl沥滤,干燥后即得到单原子Fe-N-C催化剂。通过改变金属前驱体,这种方法还可用于制备单原子的Co-N-C、Ni-N-C、Mn-N-C、FeCo-N-C、FeNi-N-C等催化剂。此外,Ohn等[54]将NiCl2与三聚氰胺的混合物高温裂解碳化后,制备了C3N4负载的单原子Ni-C-N催化剂。Deng等[55]以SiO2为模板,将(NH4)6Mo7O24与Co(NO3)2、CS2的混合物高温裂解后,制备了单原子Co掺杂三维多孔结构的MoS2催化剂。

图4 单原子Fe-N-C的制备示意图[53]Fig.4 Schematic illustration of the synthesis of single atom Fe-N-C[53]
2 非贵金属单原子催化剂的结构表征
理解单原子剂的结构及单原子催化的基本机理对于设计高性能和高稳定性的催化剂体系至关重要[56]。包括HAADF-STEM、XAFS、X-射线光电子能谱(XPS)、穆斯堡尔谱等在内的表征手段以及理论计算的发展对于单原子催化剂的发现、开发、优化及其催化机理的理解等方面具有不可替代的作用。其中,HAADF-STEM是唯一能够直接“看到”催化剂中单个原子的工具,可提供具有空间分辨的局部信息,对于含有从微米到纳米不同级别不均一性的多相催化剂而言,该技术尤其具有价值[57];包括X射线吸收近边结构(XANES)以及扩展X射线吸收精细结构(EXAFS)的XAFS则能够强有力地解析中心吸收原子的配位环境及电子态信息[58];XPS可以对中心金属的电子态信息及部分配位环境进行辅助验证;穆斯堡尔谱则可以分析Fe基单原子催化剂中Fe的超精细结构。只有将这些表征方法及其他分析手段合理地组合并与理论计算相结合,才能对非贵金属单原子催化剂的结构进行有效地表征。
如前所述,Liu等[39]以Mg(OH)2为模板通过高温裂解法制备了Co-N-C单原子催化剂,并对其结构进行了解析。图5(a)为Co-N-C的HETEM图,可以看出无明显的Co纳米颗粒,说明Co可能以原子簇或单原子的形式存在。图5(b)为HAADF-STEM图,直接证明了Co以单原子的形式高密度地分散在碳载体上。图5(c)为Co-N-C及对比样品的XANES谱图,Co-N-C在7714~7716 eV的边前区内没有吸收峰,说明Co-N-C中的CoN4位点不是平面结构。进一步利用密度泛函理论(DFT)对不同构型的CoN4的XANES谱图进行计算,从图5(d)可以看出,CoN4C8-1-2O2构型的计算值与实验表征结果最为一致。此时,Co中心与4个吡啶N成键,形成变形的平面正方形配位结构;轴向则有2个弱吸附的O2分子吸附在Co原子上。图5(e)与(f)为经过傅里叶变换的EXAFS谱图,通过与Co箔以及Co3O4的对比,可以看出Co-N-C中无Co-Co键及Co-O键,再次证明了Co的单原子分布;并且EXAFS谱图的拟合结果与实验表征结果具有一致性,验证了模型的准确性。
Liu等[40]还对以纳米MgO为模版,不同温度下高温裂解制备的单原子Fe-N-C催化剂的活性物种进行了结构解析。HAADF-STEM、EXAFS分析表明,600℃、700℃制备的Fe-N-C-600以及Fe-N-C-700中Fe物种原子分散,Fe-N-C-800中则存在少量的Fe纳米颗粒。对应的XANES谱图在7717 eV的边前区内有明显吸收峰,表明3种催化剂中FeN4为平面正方形配位结构。此外,XPS给出Fe的价态为Fe3+。图6为Fe-N-C-600/700/800的57Fe穆斯堡尔谱图[40]。从图6可以看出,Fe-N-C-600/700的谱图可由3条双峰拟合,而Fe-N-C-800的谱图由2条双峰、1条单峰、1条线峰拟合。其中,单峰与六线峰表明Fe-N-C-800中存在γ-Fe以及FexC物种。进一步根据同质异能移、四级分裂值对双峰进行分类,将上述双峰分为D1、D2、D3、D4共4种类型。D1归属于类卟啉铁FeⅡN4物种,D2为高自旋态的X-FeⅢN4-Y物种(X、Y为配位的O或N),D3为低自旋态的N-FeⅢN4-N 物种,D4则为5配位中自旋态的N-(FeⅢN4) 物种。这些结果表明,即使是单原子分散的Fe-N-C,Fe仍以多种形态分布,而有效地表征这些物种,是进一步研究其构效关系的基础。
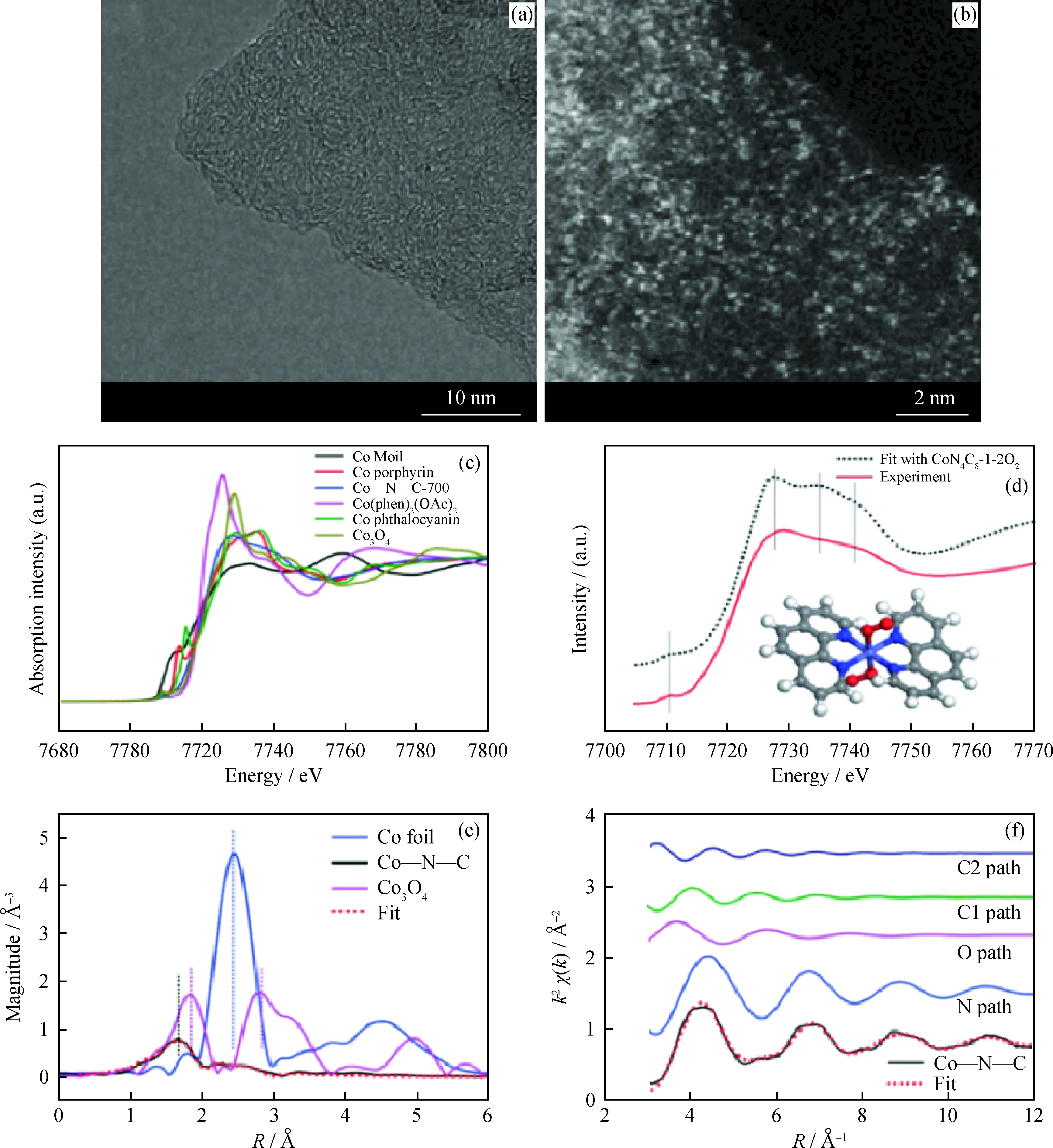
图5 单原子Co-N-C催化剂的结构解析[39]Fig.5 Structure analysis of single-atom dispersed Co-N-C catalyst[39](a) HRTEM and (b) HAADF-STEM images of Co-N-C catalyst; (c) The normalized XANES spectra at the Co K-edge of different samples and (d) Comparison between Co-N-C catalyst and the theoretical spectrum (black dotted line); (e) The k2-weighted Fourier transform spectra of different samples; (f) The contributions of different paths including Co-N (blue line), Co-O (pink line) and Co-C (green and navy bluelines) in k-space for the Co-N-C sample
六

图6 Fe-N-C-600/700/800的57Fe穆斯堡尔谱图[40]Fig.6 57Fe Mössbauer spectra of the Fe-N-C-600/700/800[40]
3 非贵金属单原子催化剂的应用
3.1 电催化反应
氧还原反应(Oxygen reduction reaction, ORR)、析氧反应(Oxygen evolution reaction, OER)和HER是电解水、燃料电池、金属-空气电池等绿色能源技术的核心,催化剂对于促进这些反应起到重要作用[59]。目前常用的ORR和HER的催化剂是Pt基材料,而OER的催化剂则是IrOx或RuOx。基于导电多孔碳载体构建的单原子Co(Fe)-N-C 催化剂,由于具有良好的耐酸碱、抗毒化、离子、电子传输性能,并且利于气相反应物、产物的扩散,被视为一种潜在的、可替代贵金属催化剂的高性能电催化材料。
Liang等[37]将裂解法制备的Co(Fe)-N-C应用于酸性条件下的ORR。其中以SiO2纳米颗粒为模板制备的Co-N-C具有较高的比表面积、均一的孔分布以及分散的Co-N-C位点,表现出最好的催化性能,ORR半波电位0.79 V,仅比20%Pt/C负移58 mV,且经循环伏安10000圈加速老化测试(AAT)后半波电位仅负移9 mV。Zhang等[52]以粒径50 nm的FeZnZIF为前驱体制备的Fe-N-C催化剂,在酸性条件下的ORR半波电位达到了0.85 V,略低于Pt/C(60 μg Pt/cm2)的0.88 V,AAT实验表明,催化剂的稳定性远优于Pt/C;此外催化剂在ORR过程中H2O2的产率小于1%,表明此反应是1个4电子还原过程。最近,Shen等[38]的研究表明,在Fe-N-C中掺入S形成C-S-C 位点,有利于提升催化剂在酸性条件下的ORR性能。密度泛函理论(DFT)计算结果表明,当S与Fe的距离大于0.73 nm时可有效减少Fe中心的电子局域,促进Fe费米能级附近电子态与ORR中间态氧物种的相互作用,从而有效降低过电位。而在碱性条件下,Yin等[49]制备的单原子Co-N-C催化剂ORR半波电位0.881 V,优于20%Pt/C的0.811 V。Chen等[51]制备的单原子Fe-N-C催化剂ORR半波电位更是达到了0.900 V,并且具有传统Pt/C催化剂所不具备的优异的抗甲醇毒化能力以及稳定性。DFT计算结果表明,单原子分散的FeN4中心有利于电子传输以及中间产物OH吸附物种转化为OH-,是催化剂具有优异性能的关键。
在OER的应用研究方面,Li等[60]的理论计算结果表明,石墨相氮化碳(g-CN)负载的单原子Co、Ni催化剂具有较好的应该前景。Ohn等[54]则制备了g-CN负载的单原子Ni并应用于OER,结果表明,负载单原子Ni的g-CN的OER性能有显著提升,但其催化过电位与IrOx催化剂仍有一定差距。Zheng等[45]的DFT计算结果表明,C3N4空穴稳定的单原子Co所形成的CoN3C2位点具有与贵金属催化剂相近的OER及ORR催化性能。基于此结果,他们制备了包覆C3N4的CNT负载的单原子Co催化剂,并在1 mol/L的KOH电解液中评价OER性能,在电流密度达到10 mA/cm2时,对应的电位1.61 V,略高于IrO2催化剂。而经过900℃高温处理破坏C3N4结构的催化剂性能明显下降,表明C3N4与Co形成的CoN3C2是实际的活性位点。此外,Chen等[44]制备的CNT负载S掺杂的单原子Fe-N-C同样是OER、ORR双功能催化剂,并且以此催化剂构建的锌-空气电池器件在功率密度以及循环性能等方面均优于对应的20% Pt/C催化剂。通过与不含S的催化剂比较,他们认为,S、N对于C骨架的电荷分布以及活性位点FeNx的3d电子密度的影响是提高催化性能的关键。
在HER的应用研究方面,Qiu等[22]制备的单原子Ni催化剂在酸性条件下表现出突出的催化性能,起始过电位约为50 mV,Tafel斜率为45 mV/dec。XPS与理论计算表明,Ni嵌入到石墨烯层内,与C之间键和并发生电子转移,形成空的杂化轨道是单原子Ni催化剂具有突出HER催化活性与稳定性的关键。Fan等以原位活化法制备的石墨烯负载单原子Ni催化剂同样具有优异的HER催化性能,Tafel斜率为41 mV/dec,在电流密度为100 mA/cm2时,过电位仅为112 mV。此外,Fei等[43]制备的N掺杂石墨烯负载的单原子Co催化剂,在酸性条件下HER起始过电位为30 mV,Tafel斜率为82 mV/dec。通过比较不同Co负载量及N掺杂量的催化剂,他们认为,活性位点是与N配位的Co中心。总体而言,对于ORR、OER以及HER,非贵金属单原子催化剂相较于贵金属催化剂在抗毒化以及稳定性方面有明显的优势。尤其在碱性条件下的ORR,非贵金属单原子催化剂的催化活性甚至优于贵金属催化剂;但对于OER以及HER,其催化活性仍需进一步提高。
3.2 加氢及脱氢反应
偶氮苯类化合物是一种广泛使用的染料及医药中间体,通常由贵金属催化剂催化加氢芳香硝基化合物制得[61]。Liu等[39]以高温裂解法制备单原子Co-N-C催化剂,并首次将非贵金属单原子催化剂应用于此类加氢反应。Co-N-C催化剂在3 MPa H2、80℃的反应条件下,即可将硝基苯转化为偶氮苯。该催化剂具有优异的底物普适性,即使苯环侧链含有-C=C,-I,-Br等可还原性基团时,也可制备相应偶氮苯衍生物,催化选择性明显优于传统的贵金属催化剂。傅里叶变换衰减全反射红外光谱表明,反应物的-C=C等官能团在Co-N-C 活性中心没有发生吸附,是其具有高选择性的主要原因。Zhu等[33]发现,单原子Cu催化剂可应用于5-羟甲基糠醛低温加氢制2,5-呋喃二甲醇,催化活性比传统沉淀法、浸渍法制备的含Cu纳米颗粒催化剂的活性高了1、2个数量级。进一步分析表明,单原子Cu在载体表面形成Cu0-Cu+协同位点。其中,Cu0主要起解离H2的作用,Cu+作为亲电位点则有利于反应物中C=O的吸附与活化。Li等[20]将NU-1000负载的单位点Ni催化剂应用于乙烯的加氢反应,其在50℃下单一Ni催化乙烯加氢的转化速率(TOF)约为0.9 s-1,并且催化剂在100℃反应14 d后仍保留90%的初始活性。此单位点Ni催化剂在氯化二乙基铝存在的条件下,还可有效的催化乙烯聚合,其TOF约为0.07 s-1,优于其他基于MOF构建的烯烃聚合催化剂。最近Dai等[50]则将单原子Ni-N-C催化剂应用于乙炔选择性加氢制乙烯,在乙炔高转化率(>90%)的条件下,乙烯的选择性仍接近90%,这一性能甚至优于贵金属Au-Pd催化剂。进一步分析表明,NiN4位点中N对单原子Ni的d电子轨道的调控,是催化剂取得高选择性的关键。
在脱氢反应的应用研究方面,Guo等[34]将Fe©SiO2应用于甲烷制乙烯、芳烃的反应中。研究表明,甲烷分子在配位不饱和的Fe中心上催化活化脱氢,获得表面吸附态的甲基物种。在传统催化剂表面,吸附态甲基物种容易进一步脱氢或者发生C-C偶联而造成积炭失活。而单位点分散的Fe由于缺少相邻的Fe-Fe位点,甲基物种容易从表面脱附形成高活性的甲基自由基,随后在气相中经自由基偶联反应生成乙烯和其它高碳芳烃分子。如图7所示,甲基物种在单位点Fe上脱附形成甲基自由基的能垒仅有2.32 eV;Fe中心吸附第2个甲烷分子并形成甲基自由基的能垒则分别为3.07和2.19 eV。在反应温度1090℃和空速21.4 L/(gcat·h)条件下,单位点Fe催化剂的甲烷单程转化率达48.1%,乙烯的选择性为48.4%,所有产物(乙烯、苯和萘)的选择性>99%,实现了CO2的零排放。此外,Li等[21]将NU-1000负载的单位点Co催化剂应用于丙烷的氧化脱氢,催化剂在反应温度200℃下即表现出了明显的催化性能。然而当丙烷转化率超过10%时,大部分丙烷发生深度氧化,丙烯的选择性不足30%,如何实现低温下选择性氧化脱氢仍是该研究领域的难点。
3.3 液相氧化反应
苯酚是重要的有机化工原料,温和条件下、高效的将苯直接羟化合成苯酚具有重要的经济意义与研究价值,是催化领域的一个研究热点[62]。Deng等[27]研究发现,FeN4/GN即使在0℃的条件下,以H2O2为氧化剂即可催化氧化苯制苯酚。而在25℃反应温度下,FeN4/GN催化苯转化的初始TOF为84.7 h-1,反应24 h后苯的转化率为23.4%,苯酚的产率为18.7%。利用DFT计算,他们认为,FeN4的中心Fe原子通过解离H2O2形成O=Fe=O,这一位点可以有效地吸附苯分子,随后发生氢转移反应将苯直接氧化成苯酚。XAFS表征结果也支持催化剂活性中心O=Fe=O的形成。Zhang等[53]同样将单原子的Fe-N-C应用于催化氧化苯制苯酚。他们认为,Fe首先将H2O2分解形成2个羟基自由基,随后生成1个水分子以及活性氧物种,活性氧物种直接将苯氧化成苯酚。除了苯的催化氧化外,Liu等[40]以叔丁基过氧化氢为氧化剂,将Fe-N-C应用于乙苯的选择性催化氧化,苯乙酮的选择性高达99%。并且该催化剂具有优异的底物普适性,即使苯环侧链含有-OCH3等给电子基团或者-NO2等吸电子基团,或者反应物为杂环化合物以及环己烷时,也可高选择性地制备相应酮类化合物。为了探究反应的活性位点,以KSCN为滴定剂对Fe中心进行选择性毒化并比较催化性能,结合穆斯堡尔谱的表征,推断中自旋态的N-(FeⅢN4)是活性最高的FeN4物种。此外,Zhang等[42]以Co-N-C为催化剂,O2为氧化剂,催化氧化一级醇与二级醇脱氢偶联,制备α,β不饱和酮。Xie等[63]以Cu(Fe, Co, Ni, Cr)-N-C 为催化剂,O2为氧化剂,催化氧化醇,制备对应的醛。

图7 Fe©SiO2在1223 K催化甲烷形成甲基自由基的能量剖面图[34]Fig.7 DFT calculations on catalytic generation of methyl radicals over Fe©SiO2 at 1223 K[34]
3.4 光催化还原CO2及其他反应
CO2的资源化再生利用,对于同时解决温室效应与能源短缺两大问题具有重要的理论价值和现实意义,其中光催化还原CO2是目前最主要的研究方向之一[64]。Zhang等[23]比较了MOF-525负载了单位点Co前后的可见光催化还原CO2性能。其中MOF-525-Co在以三乙醇胺为电子牺牲体的条件下,光催化CO2生成CO的速率为200.6 μmol/(g·h),生成CH4的速率则为36.67 μmol/(g·h)。研究表明,MOF-525的卟啉环引入单原子Co后,极大的提升了卟啉环的电子空穴分离效率,延长了光生电子的寿命并促进了吸附在Co中心的CO2的还原。Long等[30]制备的Pd7Cu1-TiO2光催化还原CO2生成CH4的速率为19.6 μmol/(g·h),选择性达到96%。理论计算与实验结果共同表明,单原子分散的Cu起到了2个关键作用:(1)形成配对的Pd-Cu位点促进了反应物CO2的吸附,并且抑制了副产物H2的生成;(2)Pd晶格调控了Cu位点的d带中心位置,有利于吸附的CO2的活化。除了光催化还原CO2外,Liu等[65]近期的研究表明,单位点的Co催化剂可以有效分离和传输光生电子和空穴,实现了高效、自发的太阳光催化全解水析氢。此外,近期的研究结果还表明,非贵金属单原子催化剂可应用于染料敏化太阳能电池[28],电催化还原CO2[66],CO的催化氧化[67-69],水煤气变换[70]等一系列重要反应。
4 结论与展望
单原子催化这一多相催化领域的新概念的提出,在一定程度上模糊了多相催化与均相催化的界限,将金属催化剂的原子利用率提高到新的高度,促进对多相催化过程与机理的认识向原子级别层面的深入。从金属催化剂自身成本的角度出发,非贵金属催化剂相对于贵金属催化剂具有天然的优势;自然界中存在的几类十分重要的高效非贵金属单原子催化剂则为非贵金属单原子催化剂的发展树立了标杆与方向。
当前,非贵金属单原子催化剂的制备与应用研究方面已取得了诸多进展,但也存在着许多难题与挑战:(1)无论是基于特定设备的ALD法、电弧放电法,或是以金属配位有机化合物为前驱体的高温裂解法、球磨法等制备的非贵金属单原子催化剂成本仍相对较高,并且在催化剂的可控制备、宏量制备方面还存在着许多不足;(2)载体的类型与结构对于单原子催化剂的性能起着重要作用,目前已成功研究的载体仅限于碳载体、硅载体、MOF等少数类型,如何扩展载体类型以提升催化性能甚至构建双功能催化剂仍有许多难点;(3)扩展Fe、Co、Ni外的非贵金属单原子的制备与应用并提升非贵金属的负载量;(4)对非金属单原子催化剂的结构解析并阐明其在特定反应中的构效关系。
非贵金属单原子催化剂的出现与发展为贵金属催化剂的替代提供了新的可能,尽管该领域目前的研究仍处于实验室阶段且存在诸多挑战,但其迅猛的发展势头与广阔的发展前景势必将推动该领域的研究不断向工业化应用方向前行。
