H-Zr-MCM-48的合成及其正庚烷异构性能
2018-08-10马守涛所艳华汪颖军
王 坚, 马守涛,2, 张 微, 所艳华, 汪颖军
(1.东北石油大学 化学与化工学院, 黑龙江 大庆 163000; 2.石油化工研究所 大庆化工研究中心, 黑龙江 大庆 163714)
作为现代社会必不可少的能源之一,汽油的使用推进了工业化的进程,但随之而来的环境问题日益凸显,中国第5阶段车用汽油国家标准(GB 17930—2013)的发布进一步表明国家对国内炼油行业的技术进步和装置改造的要求提高。且从2017年1月1日起,北京已全面推行京VI油品标准,其要求汽油苯体积分数降到0.8%以下,烯烃降到15%以下,芳烃降到35%以下,50%蒸发温度降到110℃以下,作为目前为止最严格的排放标准,使得清洁、优良的高辛烷值汽油调和组分需求更加迫切。根据这些需求,各类催化剂的研究成为焦点。如由ZSM-5分子筛改性而衍生的FCC、DCC、C4~C8烃裂化、MTP等多种增产轻烯烃技术,在SAPO-34的合成改性过程中所开发的MTO技术,在β、MCM-22介孔材料合成与应用过程中所开发的液相法乙苯技术等,因此介孔材料的开发研究具有重大意义。作为提高汽油辛烷值的重要方法,烷烃异构化的发展与应用已受到广泛关注,但近年来长链烷烃异构化催化剂主要以负载型微孔沸石双功能催化剂(ZSM-5、SAPO-11、Y、MOR)为主,该类催化剂的裂解率高且孔道狭窄,对长链烷烃向多支链异构烷烃的转化反应选择性较低。因此开发研究比表面积较大、孔隙率高、热稳定性良好、催化活性较高的催化剂成为研究者们研究的热点。
MCM-48介孔材料具有最小表面拓扑结构且镜面对称的三维螺旋面孔道结构、比表面积较大及良好的长程周期性和稳定的骨架结构,因此采用MCM-48介孔材料作为载体对物料传输和活性组分分散具有极大优势[1]。但纯硅MCM-48介孔材料合成相区较窄,酸强度低,热稳定性差[2],为了加快介孔材料的工业应用,近些年国内外研究者多专注于将具有催化作用的过渡金属均匀掺杂于纯硅介孔材料中,形成具有催化活性的杂原子分子筛催化剂。浸渍法[3]、水热合成法[4]、离子交换法[5]等方法的应用,使Sn、Cr、V、Fe、Cu、Pt、Zr等金属成功引入到MCM-48介孔材料中。引入这些杂原子进入介孔材料骨架中,介孔材料结构未发生明显改变,但可以使其酸性、粒度、孔道结构等物化性能得到改善,如Wang等[6]研究发现,氟离子能够提高硅源聚合能力,并且能够在分子筛表面形成氟化硅,其憎水性要远大于硅羟基,从而提高了分子筛的水热稳定性。笔者以硝酸锆为锆源,氟化钠为无机添加剂,制备了Zr-MCM-48介孔材料,考察了介孔材料合成过程中金属锆的引入量对Zr-MCM-48织构的影响,同时以离子交换法制得H-Zr-MCM-48催化剂,并以正庚烷异构化反应为探针考察了其催化性能。
1 实验部分
1.1 试剂与仪器
正硅酸乙酯(TEOS,AR)、十六烷基三甲基溴化铵(CTAB,AR)、硝酸锆(Zr(NO3)4·5H2O,AR)、氟化钠(NaF,GR)、氢氧化钠(NaOH,GR),天津市大茂化学试剂厂产品;正庚烷(C7H16,AR),沈阳市华东试剂厂产品;硝酸铵(NH4NO3,AR),天津市耀华化工厂产品;去离子水,东北石油大学自制。
样品物相表征采用日本株式会社理学D/Max IIB型X射线衍射仪。实验条件:CuKα光源,40 kV×20 mA,扫描速率为2°/min,扫描范围为1.5°~10°和10°~80°。N2吸附-脱附在美国康塔NOVA/2000e型比表面积和孔隙度分析仪上测定,液氮温度下的氮气作为吸附质。NH3程序升温脱附在AutochemII 2920上测定,取0.2 g样品,在20 mL/min 的氦气中升温至450℃,活化处理 0.5 h,降温至150℃,脉冲吸附氨气至饱和,用氦气吹扫样品表面物理吸附的氨气,程序升温至700℃,升温速率为10℃/min。TCD为检测器检测脱附信号。样品的孔结构在Phillips TENCNAI-12型透射电子显微镜下测得,加速电压为100~120 kV。
1.2 催化剂的制备

1.3 催化剂的性能评价
在内径为6 mm反应管的不锈钢固定床反应器中进行正庚烷异构化反应。步骤为:将0.2 g催化剂置于底部装有少量石英砂及石棉网的反应管中,在氢气流中以2℃/min的升温速率升至一定温度还原一定时间后,降温至一定温度,通入氢气和正庚烷发生反应。反应稳定30 min后开始取样分析,产物在GC-7980A型气相色谱仪上进行分析。
2 结果与讨论
2.1 合成的Zr-MCM-48和H-Zr-MCM-48催化剂的物性表征
2.1.1 XRD表征
图1为MCM-48和Zr-MCM-48-x的XRD谱。由图1可知,样品均为典型的三维立方结构[7],MCM-48 的特征衍射峰在掺杂不定量的金属Zr后均发生了不同程度的变化。当金属掺杂量较低(x≤0.06)时,在2θ=2°~4°之间可观察到(211)和(220)特征衍射峰,这说明样品的有序特征依然存在[5]。随着金属掺杂量的增加,虽然(211)特征峰依然存在,但(220)特征峰的强度逐渐下降,这可能是因为随着金属Zr含量增加,骨架有序度逐渐降低,介孔结构遭到破坏。因此,选择合适的Zr与Si的比例是合成出结构完好的Zr-MCM-48的重要条件。图2为Zr-MCM-48-0.02 的广角XRD谱图。由图2可发现,样品特征峰上只有无定形SiO2的非晶相峰,说明金属Zr高度分散在MCM-48介孔材料中,没有发生团聚[8]。图3为Zr-MCM-48-0.02与H-Zr-MCM-48-0.02 的XRD谱图对比。由图3可以明显看出,虽然H-Zr-MCM-48-0.02特征衍射峰强度降低,但三维立方结构并没有被破坏,因此仍可从介孔材料角度分析之后的实验结果。

图1 MCM-48和Zr-MCM-48-x样品的XRD谱图Fig.1 XRD patterns of MCM-48 and Zr-MCM-48-x x: (1) 0; (2) 0.02; (3) 0.04; (4) 0.06; (5) 0.10
通过观察图1可知,Zr-MCM-48介孔材料的特征衍射峰与MCM-48有两点明显变化,首先是特征峰强度降低,这是分子筛的有序度降低所导致的。其次是随着Zr含量的增加,Zr-MCM-48特征衍射峰向低2θ角移动,晶面峰d100值增大[9]。由以上两点可推测Zr可能进入了Si—O骨架,这是因为所引入金属杂原子Zr的原子半径大于Si原子半径,引入Zr后,更多较长的Zr—O键替代Si—O键,使分子筛平均孔道厚度增加,结构扭曲程度增大,进而导致分子筛有序性降低[10]。与MCM-48相比,Zr-MCM-48的XRD特征峰向高角度偏移,说明晶面峰d100值变小,即金属掺杂后介孔晶面距离要比纯硅MCM-48小。原因可能是在分子筛的合成过程中,正电模板胶束与母液中负电荷形成静电平衡的双电层,当带正电荷的Zr4+存在于溶液中时,胶束之间的距离由于表面正电荷密度增大及双电层的静电引力增强而收缩。由此分析可知,掺杂杂原子后,当无机物种在模板剂胶束上进行骨架组装时,形成的无机墙相对纯硅分子筛变薄[11]。

图2 Zr-MCM-48-0.02的广角XRD谱图Fig.2 Wide-angle XRD pattern of Zr-MCM-48-0.02

图3 Zr-MCM-48-0.02和H-Zr-MCM-48-0.02的XRD谱图Fig.3 XRD patterns of Zr-MCM-48-0.02 and H-Zr-MCM-48-0.02 (1) H-Zr-MCM-48-0.02; (2) Zr-MCM-48-0.02
2.1.2 TEM表征
图4为所合成样品MCM-48、Zr-MCM-48-0.02及H-Zr-MCM-48-0.02的透射电镜图。从图4可以看出,所合成的样品孔道结构均比较完整,表明Zr-MCM-48-0.02 样品具有有序的介孔结构,且离子交换后,H-Zr-MCM-48-0.02的孔结构也没有被破坏。

图4 样品MCM-48及Zr-MCM-48-0.02、H-Zr-MCM-48-0.02的TEM图Fig.4 TEM images of the products of MCM-48, Zr-MCM-48-0.02 and H-Zr-MCM-48-0.02(a) MCM-48; (b) Zr-MCM-48-0.02; (c) H-Zr-MCM-48-0.02
2.1.3 SEM表征
图5为MCM-48、Zr-MCM-48-0.02及H-Zr-MCM-48-0.02 的扫描电镜图。从图5可以看到,Zr-MCM-48-0.02与MCM-48的形貌相似,均为球形。其中MCM-48形状多为粒径在0.5~1 μm的不规则球形颗粒,而Zr-MCM-48-0.02和H-Zr-MCM-48-0.02 颗粒大小不均一,所形成的粒径在0.3~0.5 μm之间,且表面伴有少量结焦物质产生,分子筛颗粒间分散度降低。二者在形貌上的区别说明过渡金属Zr掺杂进入分子筛时对分子筛的结晶过程有一定影响,这与XRD分析相吻合。

图5 样品MCM-48及Zr-MCM-48-0.02、H-Zr-MCM-48-0.02的SEM图Fig.5 SEM images of the products of MCM-48, Zr-MCM-48-0.02 and H-Zr-MCM-48-0.02(a) MCM-48; (b) Zr-MCM-48-0.02; (c) H-Zr-MCM-48-0.02
2.1.4 N2吸附-脱附表征
表1列出了添加不同Zr含量的Zr-MCM-48-x样品的比表面积和孔体积测定结果。从表1看出,随着Zr掺杂量的提高,Zr-MCM-48的比表面积从1315 m2/g下降到1007 m2/g,样品的孔径则从2.3 nm 增加到2.51 nm,这进一步推测金属Zr可能进入了MCM-48骨架。当x=0.1时,孔径减小,可能是由于金属含量过高,介孔结构塌陷导致的[12]。图6是样品MCM-48、Zr-MCM-48-0.02和H-Zr-MCM-48-0.02的低温氮气吸附-脱附等温曲线。由图6可以看出,3个样品的N2吸附-脱附等温线均属于LangmuirⅣ型,即典型的介孔材料吸附曲线[13]。在低压段(p/p0≤0.2),N2分子在介孔材料孔道内由单层向多层吸附,且吸附量平缓增加;当相对压力进一步增加时,在中压段(p/p0=0.25~0.4)曲线突增,这是由于毛细管凝聚导致的,说明所得介孔材料孔径均一[14];而当p/p0进一步增加时,所出现的吸附平台是由N2分子由单层到多层吸附在分子筛外表面,且N2分子已达到饱和吸附量所导致的,这一段位置决定了样品孔径大小,且该段曲线变化的宽窄程度可以作为中孔均一性的衡量标准;在最后高压阶段(p/p0>0.4),N2吸附-脱附曲线出现H4型滞后环,这类滞后环多归因于狭缝状孔道且孔径均匀,也与孔径分布曲线吻合。由图6(b)和(c)看到,与MCM-48相比,Zr-MCM-48-0.02和H-Zr-MCM-48-0.02曲线出现压力的突跃性增大,吸附曲线台阶不明显,这表明掺杂金属进入分子筛后导致介孔材料孔径增加,孔道有序性降低[15]。由图6(c)的BJH孔径分布图可以看出,经过NH4NO3溶液处理后得到的H型样品孔径相对比较均一,孔道内部结构没有发生明显改变,只是比表面积有所降低,这与XRD谱图的结果吻合。

表1 MCM-48和Zr-MCM-48-x样品的织构性质Table 1 Textural properties of MCM-48 and Zr-MCM-48-x samples
2.1.5 NH3-TPD表征
图7为样品H-Zr-MCM-48-x的NH3-TPD曲线。根据参考文献[16],低温处160℃、208℃和高温处400℃左右的脱附峰分别对应弱酸、中强酸和强酸,且酸总量与峰面积有关[17]。160℃附近的脱附峰是表面羟基产生的,而208℃和400℃所对应的中强酸和强酸的脱附峰则是由于金属进入分子筛骨架产生的[18]。从图7可以看出,未掺杂金属Zr时,纯硅MCM-48几乎不存在NH3脱附峰,这表明纯MCM-48酸性极弱。掺杂过渡金属Zr并经过离子交换后,样品的NH3-TPD曲线在200℃附近的脱附峰,归结于H+、Zr4+取代了MCM-48骨架上的Si原子所产生的Brönsted酸性位,这说明过渡金属Zr有助于提高分子筛的酸量,H-Zr-MCM-48含一定数量的中强酸。此外H-Zr-MCM-48的酸量随Zr引入量的增加而增加,中强酸的数量越多,对烷烃异构化反应越有利。
2.2 H-Zr-MCM-48的正庚烷异构化反应机理和反应条件对其催化性能的影响
2.2.1 H-Zr-MCM-48的正庚烷异构化反应的机理分析
目前用于烷烃异构化反应的催化剂一般为双功能催化剂,这一类催化剂包括两种组分:①提供加氢-脱氢功能的金属中心;②形成正碳离子的酸中心,且在酸性中心上进行骨架异构(改变侧链的位置的传统烷基迁移机理[19]及改变支链度质子化的环丙烷机理[20]同时发生),金属中心和酸中心的相互作用使其具有异构化和裂解功能。在合成的催化剂中,负责提供加氢脱氢功能的金属组分既可以是单金属体系又可以是多金属体系[13],正碳离子在金属-酸双功能催化剂上的反应一般按照烷基迁移或质子角-角迁移进行。由于异构化及裂化作用均是通过正碳离子中间体作用下反应,且由于正碳离子的不稳定状态,在正碳离子生成后会立即发生异构化或裂解作用。正构烯烃是正构烷烃在加氢-脱氢活性中心也就是金属中心上脱氢而产生,通过在附近酸性中心上接受质子形成正碳离子,然后在加氢-脱氢活性中心进一步发生脱氢反应形成烯烃正碳离子;环丙烷正碳离子进行质子角-角转移,开环即可得到a和b叔碳离子,最后在活性中心上吸附、加氢作用生成异构烷烃[22]。通过以上分析可知,烯烃从酸性活性中心通过扩散作用连接到加氢-脱氢活性中心加氢后生成产物烷烃,因此,烯烃在酸性中心的脱附和金属活性中心的吸附作用是异构化过程中至关重要的一步。所以,通过减少异构正碳离子在酸性中心上的停留时间可抑制裂化反应的发生。

图6 MCM-48、Zr-MCM-48-0.02和H-Zr-MCM-48-0.02的 N2吸附-脱附曲线Fig.6 N2 adsorption-desorption isotherms of MCM-48, Zr-MCM-48-0.02 and H-Zr-MCM-48-0.02(a) MCM-48; (b) Zr-MCM-48-0.02; (c) H-Zr-MCM-48-0.02
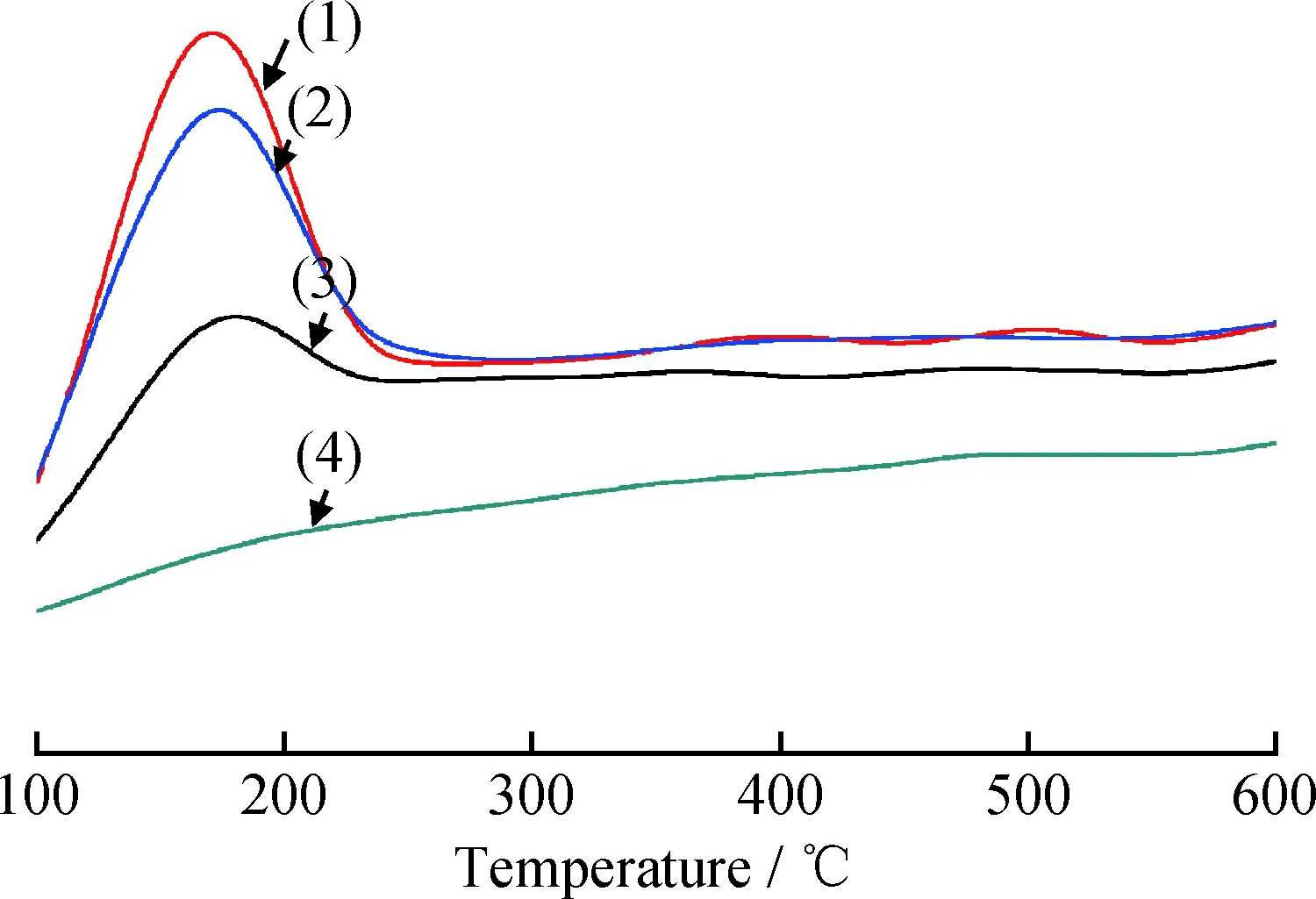
图7 H-Zr-MCM-48-x的NH3-TPD曲线Fig.7 NH3-TPD patterns of H-Zr-MCM-48-x x: (1) 0.06; (2) 0.04; (3) 0.02; (4) 0
催化剂催化性能受多种因素影响,例如催化剂晶粒大小、金属中心与酸性中心的比例、分子筛的孔结构和酸性及引入金属组分的分散情况等。至于H-Zr-MCM-48催化剂,理论上Zr4+的引入使MCM-48骨架带一定量负电荷,由于离子效应,H+会补偿一定量的骨架负电荷,从而使具有较强酸性的B酸性位Zr—OH在分子筛上形成。另一方面ZrO2与载体发生强相互作用,形成Si—O—Zr键,使催化剂表面的Zr—OH数目显著增加,出现B酸中心,且L酸中心强度增强[21]。结合之前的机理分析可知,这种强相互作用对于烷烃异构化也有促进作用。其反应过程描述如图8所示。

图8 H-Zr-MCM-48的正庚烷异构化反应的过程Fig.8 Reaction pathways of n-heptane hydroisomerization over H-Zr-MCM-48
表2为不同反应温度下H-Zr-MCM-48-0.02催化剂上正庚烷异构化反应的产物。由表2可知,产物中分别有2-MH(2-甲基己烷)、3-MH(3-甲基己烷)、2,2-DMP(2,2二甲基戊烷)、2,3-DMP(2,3二甲基戊烷)等,没有C1、C2产生。产物中并没有C7以上组分,可知该异构化反应按照单分子反应机理进行[22],且产物分布中多支链与多支链异构体的比值Rmul/mon收益在8左右,高于传统沸石催化剂[23],表明所合成的催化剂用于烷烃异构化可得到较高辛烷值的汽油组分。
2.2.2 锆/硅摩尔比(x)对H-Zr-MCM-48催化性能的影响
以正庚烷异构化反应为探针,考察了H-Zr-MCM-48-x催化剂的催化性能,结果见图9。从图9可看出,H-Zr-MCM-48-x催化剂的异构化反应活性随x的增加呈先增加后递减趋势,x=0.02时具有最高的反应活性,转化率为44.6%,而选择性则高达94.5%。根据2.2.1节中H-Zr-MCM-48催化正庚烷异构化反应的机理分析可知,分子筛的酸性质与Zr引入量成正比,但从实验中得知酸量较高的H-Zr-MCM-48-0.04催化活性反而不是最高的,出现此种现象的原因可能是由于Zr的引入量对样品孔道结构有一定改变引起的。由表1可以看出,随着Zr引入量的增加,样品的比表面积逐渐降低,离子交换后H-Zr-MCM-48的比表面积也会相应降低;而在烷烃的临氢异构化反应中,催化剂的反应活性与催化剂的酸量和比表面积均有关,因此推测,较大Zr引入量可能导致了分子筛骨架扭曲,部分未进入骨架的Zr离子与空气结合形成ZrO2堵塞分子筛的孔道,造成样品比表面积和孔体积下降,而这些外部因素在某种程度上可能会影响催化反应的传质过程,降低正庚烷与Brönsted酸性位接触的几率,从而导致催化剂的反应活性下降。
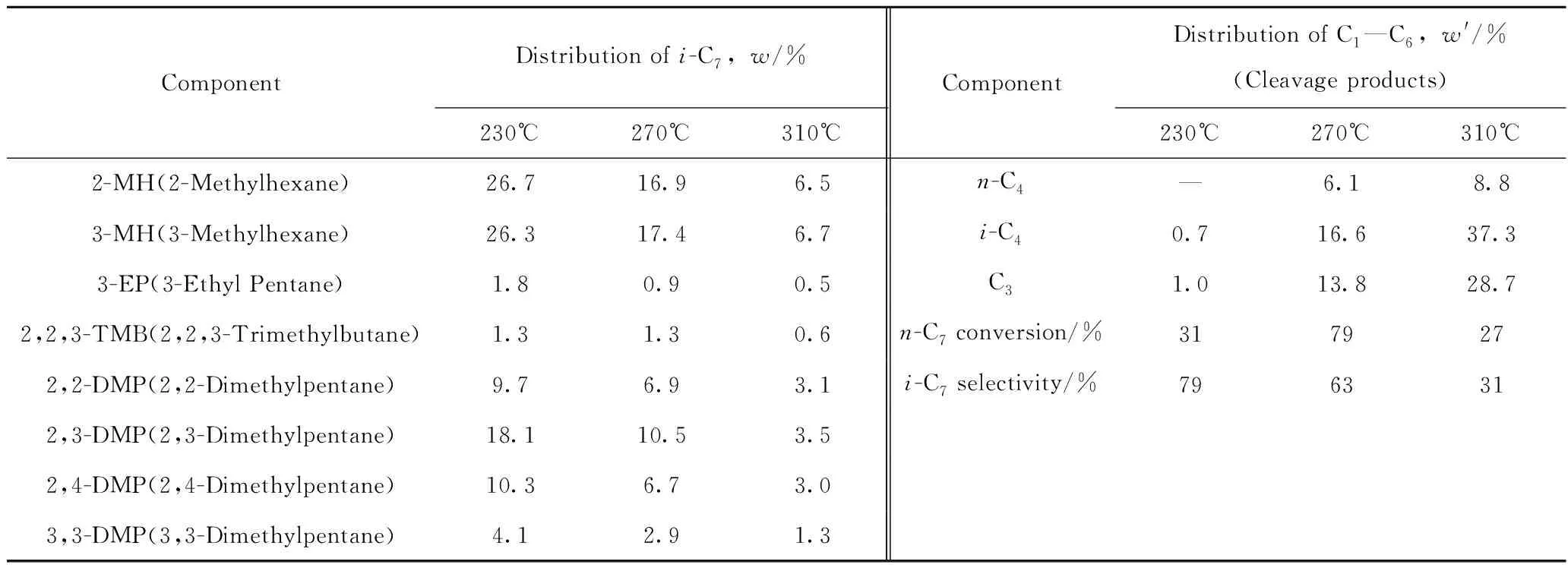
表2 不同反应温度下H-Zr-MCM-48-0.02催化剂上正庚烷异构反应的产物分布Table 2 Product distribution of n-heptane hydroisomerization over H-Zr-MCM-48-0.02 at different temperatures
w(Catalyst)=0.2 g; Reduction temperature of 400℃; Reduction time of 5 h; Reaction temperature of 230—310℃; Reaction time of 5 h

图9 H-Zr-MCM-48-x催化剂上正庚烷异构化的反应性能Fig.9 n-Heptane isomerization performance on H-Zr-MCM-48-x catalyst w(Catalyst)=0.2 g; Reduction temperature of 400℃; Reduction time of 3 h; Reaction temperature of 260℃; Reaction time of 5 h (1) n-C7 conversion; (2) i-C7 selectivity
2.2.3 反应温度对H-Zr-MCM-48催化性能的影响
烷烃异构化过程为微放热反应,而烷烃的裂解作用为吸热反应。根据热力学平衡或者阿伦尼乌斯方程可知,降低反应温度有利于异构化反应的发生,升温则有利于裂解反应的发生[24]。在较低的温度范围内,裂解反应受抑制,催化剂的选择性提高,收率增加。图10为反应温度对Zr-MCM-48-0.02和H-Zr-MCM-48-0.02催化性能的影响。由图10可以看出,对于H-Zr-MCM-48-0.02催化剂来说,反应刚开始时,正庚烷的转化率与反应温度成正比,升温至260℃时,转化率可达55.7%。低温区内(≤260℃)温度越高,催化剂活性越强。根据阿仑尼乌斯方程,Ea>0时,温度升高反应速率增加,且化学平衡向正反应方向移动,加速向热力学平衡的转化意味着转化率升高。但随着温度继续升高(>260℃),其转化率降低。导致这种情况发生有两种原因:一是反应温度过高,催化剂表面可能会发生积炭导致催化剂失活;二是可能温度升高时,裂解反应加剧,一定程度上降低了异构化产品收率[25-27]。
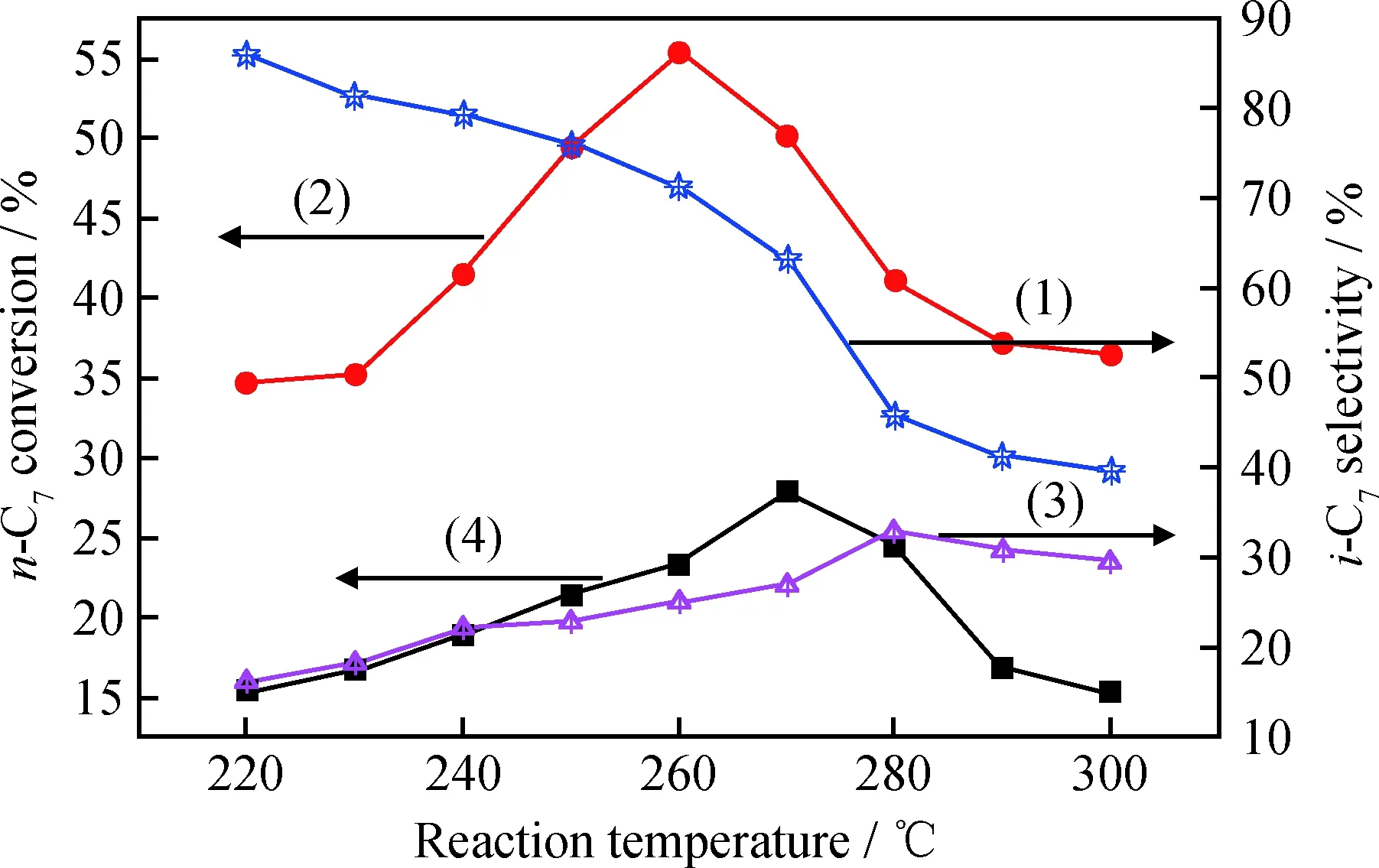
图10 反应温度对Zr-MCM-48-0.02和H-Zr-MCM-48-0.02 催化剂上正庚烷异构化反应性能的影响Fig.10 Influence of reaction temperature on the n-heptane isomerization performance of H-Zr-MCM-48-0.02 and Zr-MCM-48-0.02 w(Catalyst)=0.2 g; Reduction temperature of 400℃; Reduction time of 4 h; Reaction time of 5 h (1) The i-C7 selectivity on H-Zr-MCM-48-0.02; (2) The n-C7 conversion on H-Zr-MCM-48-0.02; (3) The i-C7 selectivity on Zr-MCM-48-0.02; (4) The n-C7 conversion on Zr-MCM-48-0.02
2.2.4 反应时间对H-Zr-MCM-48催化性能的影响
图11为反应时间对Zr-MCM-48-0.02和H-Zr-MCM-48-0.02 催化性能的影响。由图11可知,反应开始阶段2个催化剂的催化活性均随反应时间的延长逐渐升高,反应在130 min时H-Zr-MCM-48-0.02 上正庚烷的转化率和异庚烷选择性达到最大,分别为48.5%和85.7%。由前文可知,H-Zr-MCM-48-0.02酸性温和且孔道为三维螺旋结构,这种特殊结构可能使合成的催化剂具有较高的选择性。一方面,由于H-Zr-MCM-48-0.02催化剂酸性适中,在酸性位上生成碳正离子后及时脱附并在金属位加氢饱和,减少了裂化反应发生的几率。另一方面,H-Zr-MCM-48-0.02的三维螺旋孔道限制了高分子产物中间体的形成,这样就保证了H-Zr-MCM-48-0.02催化剂即使在较高转化率的条件下依然可以获得较高的异构化选择性。但在130 min 后催化活性呈下降趋势。导致H-Zr-MCM-48-0.02 催化剂在短时间内失活的原因有很多,经分析主要有如下几种可能:①活性中心上的积炭作用导致作用在金属-酸双功能催化剂上的异构化及裂解反应性能下降;②随着反应的长时间进行,一定量的金属活性相发生烧结凝聚作用,这导致金属离子不能高度分散在催化剂表面,从而降低活化作用;③还有可能是介孔材料孔道被阻塞,分子筛骨架结构坍塌或B酸位流失等[28-30]。

图11 反应时间对Zr-MCM-48-0.02和H-Zr-MCM-48-0.02 催化剂上正庚烷异构化反应性能的影响Fig.11 Influence of reaction time on the n-heptane isomerization performance of Zr-MCM-48-0.02 and H-Zr-MCM-48-0.02 w(Catalyst)=0.2 g; Reduction temperature of 400℃; Reduction time of 3 h; Reaction temperature of 260℃ (1) The i-C7 selectivity on H-Zr-MCM-48-0.02; (2) The n-C7 conversion on H-Zr-MCM-48-0.02; (3) The i-C7 selectivity on Zr-MCM-48-0.02; (4) The n-C7 conversion on Zr-MCM-48-0.02
3 结 论
(1)金属离子的引入量对介孔材料的结构有较大影响,当锆/硅摩尔比(x)低于0.6时,能合成晶型结构良好的Zr-MCM-48介孔材料,仍然保持立方相晶体结构,同时有序结构性能未发生较大改变。
(2)较之于Zr-MCM-48,在同一实验条件下,H-Zr-MCM-48催化剂展现出更好的选择性和转化率。反应温度为260℃、反应时间为130 min时催化性能最佳,正庚烷转化率为44.6%,异庚烷选择性高达94.5%。
(3)所合成的催化剂用于烷烃异构化过程,正庚烷异构化产物与理论平衡组成相比,多支链产物含量依然相对较低,因此基于提高辛烷值的要求,今后可就改变催化剂中金属酸配比进行调变,进而优化实验结果为研究方向,进行目的性研究。
