Synonymous codon usage pattern in model legume Medicago truncatula
2018-08-06SONGHuiLIUJingCHENTaoNANZhibiao
SONG Hui , LIU Jing CHEN Tao NAN Zhi-biao
1 State Key Laboratory of Grassland Agro-ecosystems, College of Pastoralagriculture Science and Technology, Lanzhou University, Lanzhou 730000, P.R.China
2 Grassland Agri-husbandry Research Center, Qingdao Agriculturaluniversity, Qingdao 266109, P.R.China
Abstract Synonymous codon usage pattern presumably re fl ects gene expression optimization as a result of molecular evolution.Though much attention has been paid to various modelorganisms ranging from prokaryotes to eukaryotes, codon usage has yet been extensively investigated for model legume Medicago truncatula. In present study, 39 531 available coding sequences (CDSs) from M. truncatula were examined for codon usage bias (CUB). Based on analyses including neutrality plots, effective number of codons plots, and correlations between optimal codons frequency and codon adaptation index,we conclude that natural selection is a major driving force in M. truncatula CUB. We have identified 30 optimal codons encoding 18 amino acids based on relative synonymous codon usage. These optimal codons characteristically end with A or T, except for AGG and TTG encoding arginine and leucine respectively. Optimal codon usage is positively correlated with the GC content at three nucleotide positions of codons and the GC content of CDSs. The abundance of expressed sequence tag is a proxy for gene expression intensity in the legume, but has no relatedness with either CDS length or GC content. Collectively, we unravel the synonymous codon usage pattern in M. truncatula, which may serve as the valuable information on genetic engineering of the model legume and forage crop.
Keywords: codon usage, gene expression, Medicago truncatula, natural selection, optimal codon
1. Introduction
The central dogma of molecular biology describes 61 codons that encode 20 amino acids in the process of translation.Fifty-nine out of the 61 codons are synonymous, i.e.,several different codons encode the same amino acid.Bias in synonymous codon usage may arise as a result of evolutionary forces such as natural selection and mutation pressure (Hershberg and Petrov 2008). Studies have shown that selection favours specific codons that promote efficient and accurate translation of genes expressed at high levels (Duret 2000; Hershberg and Petrov 2008). Thus, an effective method to identify codons favoured by selection(i.e., optimal codons) is the comparison of codon usages for each amino acid encoded by both highly and lowly expressed genes (Ingvarsson 2008; Qiu et al. 2011a; Whittle and Extavour 2015). Under mutation pressure, in contrast,preference is given to codons that tend to demonstrate lower equilibrium toward particular nucleotides (i.e., GC vs. AT) at codon positions, especially at the third nucleotide position(Sueoka 1988; Hershberg and Petrov 2008). In addition,mutation pressure can act upon genes encoding common and uncommon amino acids (Hershberg and Petrov 2008).Furthermore, many other factors can also be associated with codon usage bias (CUB). For example, base composition,relative transfer ribonucleic acid (tRNA) abundance, gene length, intron number and length, gene expression level,translation initiation efficiency, protein structure, codon and anticodon binding energy, and alternative splicing are allassociated with CUB (Rao et al. 2011; Novoa and de Pouplana 2012; Williford and Demuth 2012; Chaney and Clark 2015; Liu et al. 2015; Song et al. 2017b).
CUB in unicellular eukaryotes is relatively simple because offewer introns and fewer alternative transcripts in the organisms (Akashi 2001; Plotkin 2011). In contrast, CUB in higher plants is complex (Ikemura 1985; Ingvarsson 2007; Camiolo et al. 2012; Liu et al. 2015). Studies have demonstrated that both mutation pressure and/or selection forces are associated with CUB in various multicellular organisms (Hershberg and Petrov 2008; Plotkin 2011; Li et al. 2016). Though so far researchers have examined CUB in plants including Arabidopsis thaliana, Arachis ssp., Oryza sativa, Picea spp., Populus tremula, Silene latifolia, Triticum aestivum and Zea mays (Morton and Wright 2007; Whittle et al. 2007; Zhang et al. 2007; Ingvarsson 2010; Qiu et al.2011a; Camiolo et al. 2015; Liu et al. 2015; De La Torre et al.2015; Song et al. 2017a, c), CUB in Medicago truncatula,a model legume species, remains poorly understood.In this study, we analysed CUB in M. truncatula using 39 531 available coding sequences (CDSs). We found that natural selection is a major driving force behind CUB in M. truncatula. Furthermore, we identified 30 optimal codons for 18 amino acids, most of which characteristically end with A or T. In addition, we found that optimal codons are present frequently in CDSs with higher GC content. Thesefindings not only unravel molecular evolution in terms of synonymous codon usage in the model legume, but also serve as the valuable information on genetic improvement of the forage crop.
2. Materials and methods
2.1. Sequence data
The CDSs of M. truncatula were downloaded from the M. truncatula genome website (http://jcvi.org/medicago/display.php?pageName=General§ion=Download)(Young et al. 2011). To avoid biases caused by short sequence fragments, wefiltered the CDSs using the following criteria: (1) CDSs start with ATG and end with TAA, TAG or TGA; (2) CDSs lengths exceed 300 bp; and (3)CDSs lack premature stop codons or ambiguous codons.Data of M. truncatula tRNA abundance were obtained from GtRNAdb (version 3.0, http://gtrnadb.ucsc.edu).
2.2. Codon bias indices
We calculated the content of each of the four bases at the third synonymous codon positions (i.e., A3s, C3s, G3s, and T3s), GC content at the third synonymous codon positions(GC3s), codon adaptation index (CAI), effective number of codons (ENC), frequency of optimal codons (Fop), and relative synonymous codon usage (RSCU) using the Codon W Program (version 1.4, http://codonw.sourceforge.net).
CAI and Fop are directional measures of CUB, which quantify the degree of selection acting upon a gene (Ikemura 1985; Sharp and Li 1987). CAI values are between 0 and 1.Values close to 1 suggest that a given gene has experienced increasing intense selection to maintain a specific codon optimized for efficient translation (Sharp and Li 1987).ENC is a non-directional measure depending upon the nucleotide composition of genes. ENC values range from 20 to 61. Twenty indicates that one codon is exclusively used to encode a given amino acid, whereas 61 indicates all codons are used equally (Wright 1990). RSCU values greater than 1 indicate that a particular codon is used more frequently than expected, while RSCU values less than 1 indicate that a codon is used less frequently than expected(Sharp and Li 1987).
CDS and genomic DNA sequence lengths and the GC content at the first (GC1), second (GC2), and third (GC3)nucleotide positions of codon were calculated using an inhouse Perl script.
2.3. Data analyses
Many methods that identify optimal codons have been reported (Hershberg and Petrov 2009; Wang et al. 2011).However, RSCU value calculation is particularly popular for assessing optimal codons (Whittle et al. 2011; Whittle and Extavour 2015). Optimal codons are defined as described by Whittle and Extavour (2015). Briefly, the CDS sequences were sorted based on ENC value, and the top and bottom 5% of sequences were defined as the genes with high and low expression, respectively. ΔRSCU equals RSCUmeanhighlyexpressedCDSminus RSCUmeanlowlyexpressedCDS. A statistically significant and positive ΔRSCU value indicates that more than one codon matches this criterion per amino acid; the codon with the largest ΔRSCU for a particular amino acid is defined as the primary optimal codon (Ingvarsson 2008;Whittle et al. 2011).
A totalof 256 975 M. truncatula expressed sequence tag(EST) sequences were downloaded from the National Center for Biotechnology Information (NCBI) database on January 19, 2016. EST abundance has been used to estimate gene expression intensity (Ohlrogge and Benning 2000). In this study, we surveyed allavailable EST sequences for each M. truncatula CDSs using a local BLAST program (Altschul et al. 1997). The number of EST sequences that match a specific CDS defines the expression intensity of a given gene (Ohlrogge and Benning 2000; Song and Nan 2014).The following evaluation criteria were used as thresholds to determine sequences subjected to further analyses (Song et al. 2015): (1) length of aligned sequences>200 bp; (2)identity>96%; and (3) E-value≤10-10.
Correlation analyses were carried out using JMP 9.0(SAS Institute Inc., Cary, NC, USA), and results were depicted using Origin 9.0 (OriginLab, Northampton, MA,USA).
3. Results
3.1. Base composition of M. truncatula
A totalof 62 319 CDSs have been previously identified in the sequenced M. truncatula genome (Young et al. 2011). The total number of CDSs used in the present study was reduced to 39 531 using thefiltering criteria described in Materials and methods. The GC content in these CDSs varied from 23.9 to 69.7% (SD=3.77, Appendix A). Moreover, the GC contents at three nucleotide positions of codons were different. The GC1 was the highest (47.6%), followed by that of GC2 (38.5%) and GC3 (36.3%). The average GC contents across the three positions were 40.9%, indicating that CDSs in M. truncatula have higher AT content (59.1%).
RSCU is expressed as the observed frequency of a codon divided by its expected frequency. Thirty-one codons were less frequently used than expected (i.e., RSCU<1) and 26 codons were used more frequently (i.e., RSCU>1) in CDSs of M. truncatula, indicating the 26 codons are used preferentially among all CDSs M. truncatula (Appendix B).Furthermore, the RSCU analysis demonstrated that CDSs in M. truncatula are biased towards codons ending with A or T, except for AGG (Arg) and TTG (Leu) (Appendix B).
3.2. Factors associated with codon usage in M. truncatula
A significant correlation between the average of GC1 and GC2 (GC12) and GC3 with a slope value close to 1 suggests that mutation pressure is the major force in shaping codon usage pattern (Sueoka 1988). If natural selection is the dominant factor, in contrast, the slope value is close to 0(Sueoka 1988). In this study, a significant positive correlation(r=0.12, P<0.01, and slope=0.08) between GC12 and GC3 with a slope close to 0 was observed (Fig. 1), suggesting that natural selection has shaped the codon usage pattern in M. truncatula.
Gene spots occur along the curves of ENC plots if codons are constrained by neutral pressure (Wright 1990; Zhang et al. 2007). Other pressures in fluence codon usage if all gene spots occur below or above the ENC curve. In addition,Kawabe and Miyashita (2003) demonstrated that natural selection shapes codon usage if the GC3s across genes is narrow. Our analysis showed that in M. truncatula, most genes analysed fell below the ENC curve and GC3s values were distributed within a narrow range (0.2-0.5, Fig. 2),suggesting that natural selection plays a substantial role in the codon usage pattern.
A comparison between ENC and CAI can assess the relationship between the nucleotide composition and the natural selection (Sharp and Li 1987; Wright 1990). In this study, we found ENC and CAI had no correlation (r=0.06) in M. truncatula. There were significantly negative correlations between ENC and either A or T content at synonymous third codon positions (A3s, r=-0.26, P<0.01; and T3s, r=-0.37,P<0.01), and significantly positive correlations between ENC and either C or G content at synonymous third codon positions (C3s, r=0.40, P<0.01; and G3s, r=0.24, P<0.01).These patterns indicated that in M. truncatula, CUB was featured with high A3s and T3s (AT3s) or low G3s and C3s(GC3s) values. The reasonable explanation is that natural selection acts on the third codon position to increase the A and T content (AT3, 63.7%), instead of the G and C content(GC3, 36.3%).
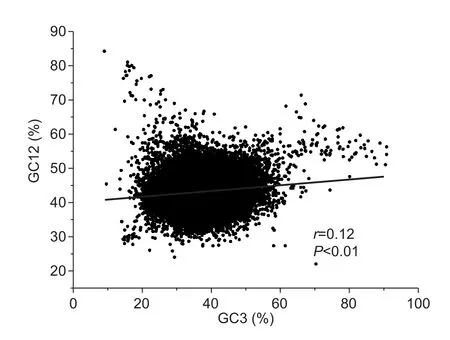
Fig. 1 Correlation between GC12 (GC1 and GC2) and GC3.GC content at the first (GC1), second (GC2), and third (GC3)codon positions were calculated using an in-house Perl script.Correlation analyses were executed in JMP 9.0, and thefigure was generated using Origin 9.0.
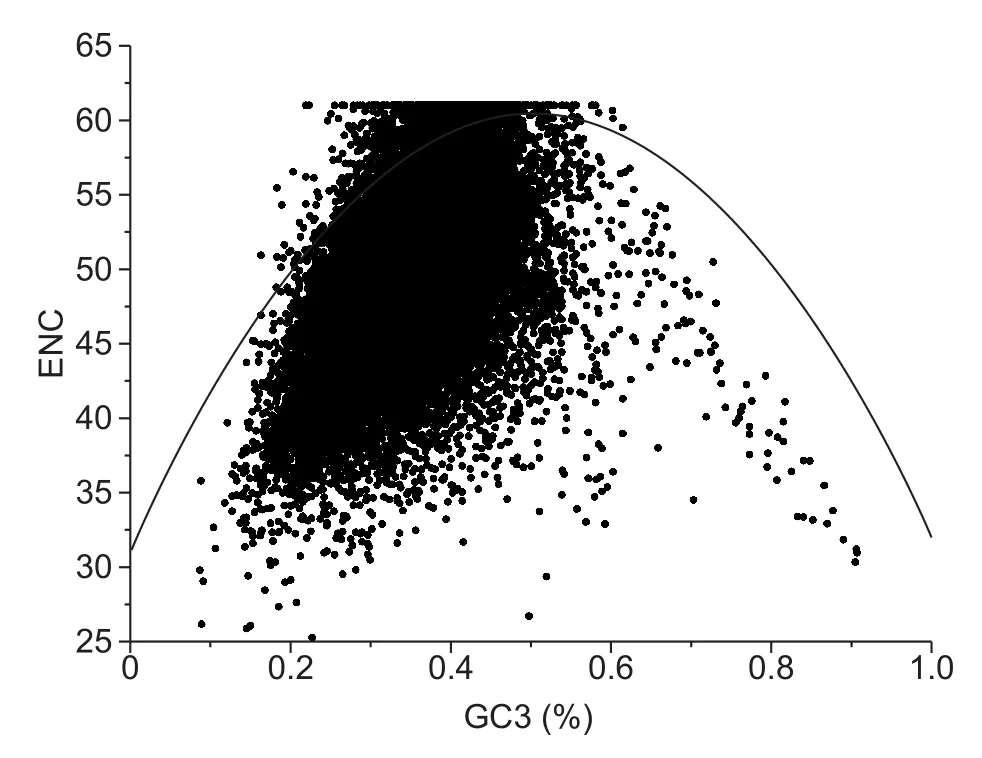
Fig. 2 Effective number of codons (ENC) plot. The ENC values shown in this plot were generated using codon W. Thefigure was generated using Origin 9.0. The continuous curve indicates the relationship between ENC and GC3s values under neutral selection. Each dot indicates a gene. GC3, GC content at the third position of synonymous codons.
3.3. ldentification of optimal codon
We identified 30 optimal codons that encode 18 amino acids in M. truncatula using the ΔRSCU method (Table 1).These optimal codons, except AGG (Arg) and TTG (Leu),preferentially end with A or T. This is consistent with the RSCU result. Furthermore, RSCU and optimal codons analyses led to the identification of 26 optimal codons with high frequency and 4 optimal codons without high frequency, whereas 28 codons are neither high frequent nor optimal.
To define the factors that determine optimal codons, we selected the Fop as an evaluation index for a correlation analysis. There was no correlation between Fop and CDS length and genomic DNA (exon and intron) length(Table 2). Significant positive correlations were observed between Fop and the GC content from different CDS and genomic DNA sequences (Table 2). These results indicated that the GC content across the three codon positions had similar effects on optimal codon usage. Moreover, optimal codon usage is associated with higher GC content in CDSs and intronic (Table 2). There was a significant positive correlation between Fop and CAI (r=0.76, P<0.01). High CAI values indicate high levels of gene expression. Thisfinding is consistent with previous studies, which showed that optimal codons were used in highly expressed genes under the impact of natural selection (Ingvarsson 2007; Qiu et al. 2011a). In general, low ENC values indicate CUB.In this study, Fop and ENC exhibited a significant positive correlation (r=0.21, P<0.01), indicating that highly optimal codons have low CUB.
3.4. Correlation between gene length, GC content and gene expression
Various factors have been examined for their association with gene expression, including GC content, intron size,and protein sequence length (Rao et al. 2011; Williford and Demuth 2012; De La Torre et al. 2015). Based on EST data analysis, gene expression intensity in M. truncatula was not correlated with CDS and genomic DNA length, and the GC content of both CDS and genomic DNA sequences (Table 2).The results indicate that CDS length and the GC content do not in fluence the gene expression in M. truncatula.
4. Discussion
As far as codon usage study is concerned, plants have remained well behind prokaryotic models. One major reason is the limited number of completely sequenced genomes in plants comparatively. Hordium vulgare,Nicotiana tabacum, Pisum sativum, T. aestivum, and Z. mays were pioneeringly investigated for their codon usage utilizing their EST or partial genome sequences(Fennoy and Bailey-Serres 1993; Kawabe and Miyashita 2003). Following the completion of A. thaliana genome sequencing in 2000, other plant genome sequences have also become available increasingly. So far codon usage has been analysed for several sequenced model plants including A. thaliana, Brachypodium distachyon, and O. sativa (Morton and Wright 2007; Qiu et al. 2011b; Liu et al. 2015). However, codon usage patterns in M. truncatula remain unexamined. In this study, we analysed codon usage patterns in M. truncatula utilizing 39 531 CDSs. Our results suggest that: (1) natural selection acts on codon usage pattern in M. truncatula; (2) for 18 out of 20 amino acids, the optimal codons characteristically end with A or T;(3) optimal codons are more widely present in genes with higher GC content; and (4) no correlation between gene expression intensity and either gene length or GC content.
In Populous and Arabidopsis, tRNA abundance is positively correlated with optimal codon usage (Wright et al. 2004; Ingvarsson 2007). However, we found that nine optimal codons (TTT, TAT, CAT, AAT, AAA, GAT, AGT,TGT and GGT) with high RSCU are associated with the low abundance of corresponding tRNA in M. truncatula (Table 1).Williford and Demuth (2012) explained this phenomenon through two hypotheses: (1) Codon-anticodon recognition heavily depends on post-transcriptional modifications of tRNA sequences. It has been confirmed that nucleotide A is always modified into I (inosine), and nucleotide U at the first anticodon position experiences extensive changes that could expand or restrict the number of recognized codons(Agris et al. 2007); (2) codons that correspond to highlyabundant tRNA cannot be translated most accurately based on 73 bacterial genomes from 20 different genera (Shah and Gilchrist 2010). Besides these hypotheses, other factors may also explain these results. First, optimal codons with low tRNA abundance may encode conserved domains.Purifying selection acts on codons with low-abundent tRNAs, many of which encode conserved domains that play a crucial role in physiological development (Zhou et al.2013; Chaney and Clark 2015). As such, proteins encoded by genes with low-abundant tRNAs have experienced purifying selection, and these proteins may play a vital role in M. truncatula. Secondly, codons with low-abundant corresponding tRNAs may be used in more frequency.In vivo analyses in Saccharomyces cerevisiae indicated that codons preferentially used in highly expressed genes are not translated faster than those highly expressed genes with non-optimal codon usage (Novoa and de Pouplana 2012;Qian et al. 2012).

Table 1 Optimal codons and transfer ribonucleic acid (tRNA) abundance in Medicago truncatula1)
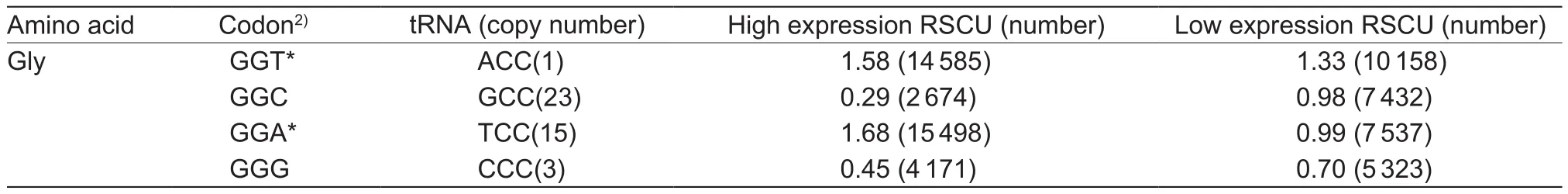
Table 1 (Continued from preceding page)

Table 2 Correlation analysis between coding sequence architecture features and gene expression based on expressed sequence tag (EST) abundance in Medicago truncatula1)
Recent studies have focused on identifying factors that act on CUB, but some resulting conclusions are inconsistent. In this study, we performed correlation analyses between Fop and a number of variables, including sequence length, GC content, CAI, and ENC. We found that optimal codon use(i.e., Fop) is not correlated with CDS length, but positively correlated with the GC content and CAI. However, Wang and Hickey (2007) found that CUB is negatively correlated with gene length, and that short genes have high GC content compared to long genes in rice. Ingvarsson (2007) showed that Fop values are negatively correlated with protein lengths, but strongly and positively correlated with the GC3 content in P. tremula. Note that CAI, which indicates CUB in genes with high expression levels, is the major factor associated positively with optimal codon usage. A strongly positive correlation has been found between CUB and gene expression in many species, including Cardamine spp.,P. tremula, S. latifolia, and Tribolium castaneum (Ingvarsson 2007; Qiu et al. 2011a; Ometto et al. 2012; Williford and Demuth 2012). When average gene expression intensities within a given tissue type are examined, Fop is not correlated with gene expression; however, when maximal gene expressions across tissues are under survey, Fop is weakly correlated with gene expression (De La Torre et al. 2015).
In M. truncatula, gene expression is not correlated with CDS length and GC content. Similar results were also observed previously in rice. Liu et al. (2004) confirmed that natural selection is one major driving force behind gene expression level, whereas CDS length only plays a minor role in rice. However, Qiu et al. (2011a) found that gene expression is positively correlated with the GC3 content, but strongly and negatively correlated with the intron GC content. GC3 is not positively correlated with gene expression in A. thaliana and A. lyrata, but there is a weak positive correlation between gene expression and intron GC content (Wright et al. 2004). The studies of the correlation between CDS or genomic DNA length and gene expression have led to controversial results. Long gene sequences actually improve gene expression in species such as T. castaneum and Picea spp. (Williford and Demuth 2012; De La Torre et al. 2015). By contrast, Camiolo et al.(2015) confirmed that short and higher-GC DNA sequences are always positively correlated with gene expression and optimalusage bias in four monocots, 15 dicots and two mosses. Some studies have proposed that short proteincoding sequences with high expression levels are less costly in terms of metabolism (Williford and Demuth 2012; Whittle and Extavour 2015). However, Yang (2009) argued that short sequences with high expression levels hardly support the energy-cost hypothesis, but may be better reconciled with the time-cost hypothesis, in which rapidly rather than highly expressed genes, tend to be expressed well in timecost efficient manner.
5. Conclusion
Our results support that nature selection played a pivotal role in forming codon usage pattern, and no relatedness between gene expression and either CDS length or GC content in M. truncatula. In addition, we have identified a totalof 30 optimal codons for the model legume. These results could provide valuable information on genetic engineering of the model legume and forage crop.
Acknowledgements
We thank Dr. Wen Jiangqi (Noble Research Institute, USA)and Dr. Wang Hongliang (United States Department of Agriculture-Agricultural Research Service, Tifton) for critical reviews and comments. This study was supported by the National Basic Research Program of China (2014CB138702)and the National Natural Science Foundation of China(31502001).
Appendices associated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
杂志排行

Journal of Integrative Agriculture的其它文章
- ldentification and characterization of Pichia membranifaciens Hmp-1 isolated from spoilage blackberry wine
- Implications of step-chilling on meat color investigated using proteome analysis of the sarcoplasmic protein fraction of beef longissimus lumborum muscle
- Spatial-temporal evolution of vegetation evapotranspiration in Hebei Province, China
- Design of a spatial sampling scheme considering the spatialautocorrelation of crop acreage included in the sampling units
- Comparison of forage yield, silage fermentative quality, anthocyanin stability, antioxidant activity, and in vitro rumen fermentation of anthocyanin-rich purple corn (Zea mays L.) stover and sticky corn stover
- The biotypes and host shifts of cotton-melon aphids Aphis gossypii in northern China