稳定同位素标记1-(3-甲氧基苯基)-[4-(4-氟-2-甲氧基-D3苯胺)甲基]-1,2,3-三氮唑的合成
2018-07-04徐仲杰涂亚辉钟佳琪汪忠华王浩然白少飞吴范宏
徐仲杰,涂亚辉,钟佳琪,孙 雯,汪忠华,王浩然,白少飞,吴范宏
(1.上海化工研究院有限公司 国家同位素工程技术研究中心 上海分中心,上海 200062;2.上海化工研究院有限公司 上海稳定性同位素工程技术研究中心,上海 200062;3.上海应用技术大学,上海 201418)
目前,各种癌症严重威胁人类生命,研发高效的抗癌药物已刻不容缓。早期的抗肿瘤药物,对肿瘤细胞和正常细胞缺乏选择性,在杀死肿瘤细胞的同时也杀死正常细胞。近年来,新发现的分子靶向药物,能够针对性地作用于肿瘤细胞,疗效良好,不良反应少,逐渐成为抗肿瘤药物的发展方向。1997年,全球第一个靶向抗肿瘤药物——利托普希单抗上市,药物研究已进入精准医疗时代,全球每年都有多个面向不同作用靶点的抗肿瘤药物进入全球医药市场。
20世纪70年代,美国科研人员在非洲灌木矮柳树的树皮中分离出天然产物——考布他汀(Combretastatin A4, CA4)。并研制出分子靶向药物,能够很好的抑制微管蛋白聚合和分裂[1]。由于其良好的生物活性,英国、美国等已将其引入临床评估,同时越来越重视对CA4结构的修饰以及类似物的开发,针对CA4结构的修饰主要集中在3个部位:A环、B环和顺式双键,成为抗肿瘤新药研究的热点方向之一[2-12]。
汪忠华等[13-14]报道了一种以苯胺丙炔和叠氮衍生物反应制备1,2,3-三氮唑衍生物的方法,具有一定的抗肿瘤活性。碳氘键比碳氢键更稳定,有利于延长药物在体内的代谢时间,提高药效,而氟代药物分子小,相容性高,本课题组利用氘标记合成的优势,在前期分子活性结构的基础上,引入氘标记的-OCD3基团,形成氘代和氟代的协同作用,创制新型氘代氟代氨甲基三氮唑类抗肿瘤新药。
1 主要仪器与试剂
IKA C-MAG HS7加热磁力搅拌器:德国IKA公司;RE-52A型旋转蒸发仪:上海卫凯仪器有限公司;BrukerSpectrospin 500 UltrashieldTM核磁共振仪:德国Bruker公司;Waters Acquity UPLC H-Class超高效液相色谱:美国Waters公司;TSQ Quantum Acess液质联用仪:美国热电公司。
碘甲烷-D3:丰度99.0%,上海化工研究院有限公司;5-氟-2-硝基苯酚:分析纯(AR),上海阿拉丁试剂有限公司;3-甲氧基苯胺、3-溴丙炔:AR,上海泰坦科技股份有限公司;浓盐酸、亚硝酸钠、甲醇、异丁醇、抗坏血酸、硫酸铜:均为AR,中国医药集团上海化学试剂公司。
2 实验方法
化合物的合成采用逆合成分析,结合有机合成原理,以稳定、廉价、易得的碘甲烷-D3为原料,经过甲基化、硝基还原、炔基取代、叠氮化以及环化反应合成1-(3-甲氧基苯基)-[4-(4-氟-2-甲氧基-D3苯胺)甲基]-1,2,3-三氮唑。合成路线示于图1。
2.1 5-氟-2-硝基苯甲醚-D3(2)的合成
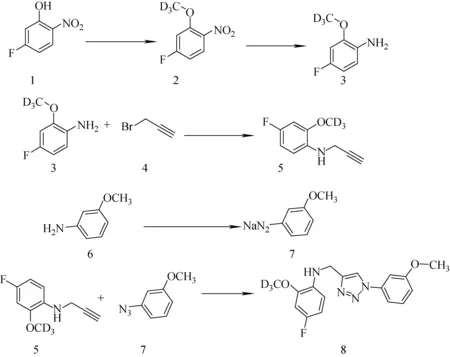
图1 化合物(8)的合成路线Fig.1 The synthetic route of compound (8)
在100 mL三口反应瓶中加入化合物5-氟-2-硝基苯酚3.4 g、无水碳酸钾5.0 g和无水丙酮40 mL,氮气置换三次,冰浴搅拌10 min后,用注射器缓慢加入碘甲烷-D33.2 g,继续冰浴反应0.5 h,然后升温回流6 h停止反应,水洗,用乙酸乙酯萃取,无水硫酸钠干燥,抽滤,浓缩,得到黄色固体5-氟-2-硝基苯甲醚-D3(2),收率90.0%(以碘甲烷-D3计)。1H NMR(CDCl3,500 MHz),δ 6.75(m,2H),7.95(dd,J1=9.0 Hz,J2=6.0 Hz,1H),鉴定为目标产物。
2.2 4-氟-2-甲氧基苯胺-D3(3)的合成
在100 mL圆底烧瓶中加入自制的5-氟-2-硝基苯甲醚-D33.5 g,加入50 mL乙醇溶解,搅拌。称取2.0 g的五水合硫酸铜溶于30 mL水中,加入至上述体系中,冰水浴下,分批向反应体系中加入硼氢化钠3.8 g,继续在冰水浴条件下反应3 h,用氯仿萃取三次,水洗,无水硫酸钠干燥,抽滤,减压去除溶剂得到棕色液体2.8 g,收率为90.5%(5-氟-2-硝基苯甲醚-D3计)。ESI-MS:145.09(M+1)+,1H NMR(CDCl3,500 MHz),δ 3.59(br,2H),6.47~6.61(m,2H),7.25(s,1H),鉴定为目标产物。
2.3 4-氟-2-甲氧基-1-丙炔基苯胺-D3(5)的合成
在100 mL三口烧瓶中加入无水碳酸钾7.0 g和5-氟-2-硝基苯胺2.8 g,加入50 mL丙酮搅拌均匀,在常温条件下滴加溴丙炔2.0 g,滴加结束后升温至50 ℃反应4 h,停止反应。水洗后用氯仿萃取,无水硫酸镁干燥,抽滤,减压除溶得黑棕色粗品。柱层析提纯后得到棕色液体2.9 g,收率85.7%(以4-氟-2-甲氧基苯胺-D3计)。ESI-MS:182.1(M)+,1H NMR(CDCl3,500 MHz),δ2.21(t,J=2.4 Hz,1H),3.94(d,J=1.8 Hz,2H),4.26(br,1H),6.56~6.62(m,3H),鉴定为目标产物。
2.4 间甲氧基苯叠氮化钠(7)的合成
将10 mL水和10 mL盐酸先后加入100 mL三口烧瓶中,冰水浴冷却后,加入3-甲氧基苯胺2.5 g,搅拌5 min,加入含亚硝酸钠1.6 g的水溶液,称取叠氮化钠1.6 g溶于水中,缓慢地滴加到反应体系中,加完后继续搅拌1 h,薄层色谱检测反应进行直至原料反应完全。萃取,洗涤,干燥,抽滤,负压除溶剂后得到间甲氧基苯叠氮化钠3.1 g,收率85.1%(以间甲氧基苯胺计)。
2.5 1-(3-甲氧基苯基)-[4-(4-氟-2-甲氧基-D3苯胺)甲基]-1,2,3-三氮唑-D3(8)的合成
在100 mL三口烧瓶中依次加入间甲氧基苯叠氮化钠1.2 g,4-氟-2-甲氧基-1-丙炔基苯胺-D31.3 g,五水合硫酸铜0.15 g,抗坏血酸钠1.2 g,异丁醇10 mL,水10 mL,室温搅拌2 h,停止反应。采用4-氟-2-甲氧基-1-丙炔基苯胺-D3后处理方法得到橘色固体粗品,柱层析得到橘色固体2.9 g,收率86.3%(以4-氟-2-甲氧基-1-丙炔基苯胺-D3计),总收率60.2%(以碘甲烷-D3计)。纯度99.0%,丰度99.0%。ESI-MS:332.11(M+1)+,1H NMR(CDCl3,500 MHz),3.86(s,3H),4.52(d,J=1.8 Hz,2H),4.54(br,1H),6.57~6.62(m,3H),6.92(s,H),7.22~7.40(m,3H),7.87(s,H)。
3 结果与分析
综上分析,在化合物1-(3-甲氧基苯基)-[4-(4-氟-2-甲氧基-D3苯胺)甲基]-1,2,3-三氮唑的(8)的五步反应过程中,产物经鉴定为目标产物。第三步合成4-氟-2-甲氧基-1-丙炔基苯胺-D3的收率初期探索时仅约50%,副产物多且影响下一步反应,其他几步合成收率稳定,基本都大于85%,故第三步反应的收率对最终产品(8)的收率具有较大影响,对第三步反应的温度、时间、投料比等条件进行优化。
3.1 合成收率影响因素
3.1.1反应温度
反应温度对4-氟-2-甲氧基-1-丙炔基苯胺-D3的收率影响较大,实验中固定反应时间4 h,n(4-氟-2-甲氧基苯胺):n1(溴丙炔)=1∶1.2,研究温度变化对收率的影响,结果示于图2。从图2结果可知,当反应体系的温度低于0 ℃时反应收率较低,其原因可能是温度低,反应活性低,反应较难发生,收率低;当温度高于50 ℃时,收率随着温度升高而降低,是因为温度高容易发生溴丙炔的二取代反应,质谱上显示有二取代的分子离子峰,并且液相上明显二取代杂峰增大,造成收率低,因此优选反应温度为50 ℃。

图2 反应温度对4-氟-2-甲氧基-1-丙炔基苯胺-D3收率的影响Fig.2 Effect of temperature on synthesis of (4-Fluoro-2-methoxy-phenyl)-prop-2-ynyl-amine-D3
3.1.2反应投料比
为了提高4-氟-2-甲氧基-1-丙炔基苯胺-D3的反应收率,研究4-氟-2-甲氧基苯胺-D3和溴丙炔的物质的量比对4-氟-2-甲氧基-1-丙炔基苯胺-D3反应收率的影响,具体结果列于表1。由表1结果可知,当反应物质的量比达到1∶1.2时,4-氟-2-甲氧基-1-丙炔基苯胺-D3的收率达到最大。

表1 反应投料比对4-氟-2-甲氧基-1-丙炔基苯胺-D3收率的影响Table 1 Effect of ratio on the synthesis of (4-Fluoro-2-methoxy-phenyl)-prop-2-ynyl-amine-D3
注:反应条件:反应时间4 h,反应温度50 ℃;n∶n1=4-氟-2-甲氧基苯胺∶溴丙炔
3.1.3反应时间
固定反应物料比为1∶1.2,反应温度为50 ℃,考察反应时间对合成4-氟-2-甲氧基-1-丙炔基苯胺-D3的影响,实验结果示于图3。由图3结果可以看出,随着反应时间的增加,4-氟-2-甲氧基-1-丙炔基苯胺-D3收率迅速提高,到4 h后基本趋于稳定,再延长时间,收率略有下降,其原因是反应时间过长导致溴丙炔二取代副产物的生成(质谱上显示有二取代的分子离子峰,并且液相上明显二取代杂峰增大),综合成本考虑,选择较优的反应时间为4 h。
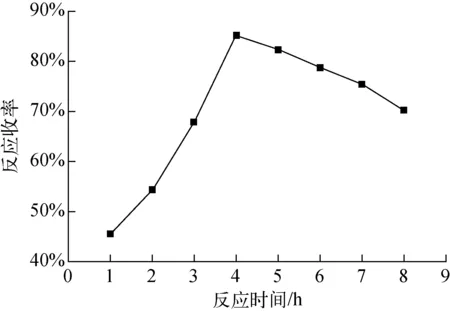
图3 反应时间对4-氟-2-甲氧基-1-丙炔基苯胺-D3合成收率的影响Fig.3 Effect of reaction time on synthesis of (4-Fluoro-2-methoxy-phenyl)-prop-2-ynyl-amine-D3
3.2 结构表征
化合物(8)的1H NMR核磁谱图分析示于图4。1H NMR(CDCl3,500 MHz),δ3.82(s,3H),3.84(s,3H),4.52(d,J=1.8 Hz,2H),4.54(br,1H),6.57~6.62(m,3H),6.92(s,H),7.22~7.40(m,3H),7.87(s,H)。谱图积分为17个氢,结构上为17个氢,可见,合成产品的1H NMR与化合物(8)的结构吻合,确定产品是目标产物。
化合物(8)的LC-MS图谱示于图5,主峰m/z=332.11,为[M+H]峰,其质量数332.11≈331.10+1(331为化合物(8)相对分子质量),结合核磁谱图可以确认为目标化合物(8),同位素丰度为99.0%。

图4 化合物(8)的1H NMR谱图Fig.4 1H NMR of compound (8)
3.3 活性测试
采用7sea-cell Counting Kit-8(CCK-8)染色法检测化合物(8)的细胞活力。取活细胞比例达90%以上的细胞进行实验。细胞增殖抑制试验采用CCK-8细胞活力检测试剂盒。细胞消化、计数、制成浓度为1×105个/mL的细胞悬液,96孔板中每孔加入100 μL细胞悬液(每孔1×104个细胞);96孔板置于37 ℃,5% CO2培养箱中培养24 h;每孔加入100 μL含药物的培养基,设立阴性对照组,溶媒对照组,阳性对照组,每组5复孔;96孔板置于37 ℃,5% CO2培养箱中培养72 h后;每孔加入10 μL CCK-8溶液,将培养板在培养箱内孵育4 h,用酶标仪测定在450 nm处的OD值,计算化合物(8)对人肺癌细胞A549、人肝癌细胞HepG2、人胃癌细胞MGC-803、人胃癌细胞MKN45等细胞的抑制率及IC50值。剂量设置:剂量1组:100 μmol/L;剂量2组:50 μmol/L;剂量3组:25 μmol/L;剂量4组:12.5 μmol/L;剂量5组:6.25 μmol/L;剂量6组:3.125 μmol/L;剂量7组:1.562 5 μmol/L;剂量8组:0.781 25 μmol/L;剂量9组:0.390 625 μmol/L;剂量10组:0.195 312 5 μmol/L;阳性对照:10 μg/mL。

图5 化合物(8)的LC-MS谱图Fig.5 The LC-MS ofcompound (8)
化合物(8)对肿瘤细胞的IC50值列于表2,表2结果表明,化合物(8)对A549、MGC803、MKN45、HepG2等表现出广谱的抗肿瘤活性,具有成为抗肿瘤药物的潜力。

表2 化合物(8)对肿瘤细胞的IC50值Table 2 The IC50 values of compound (8) to tumour cell
4 结论
本研究采用碘甲烷-D3为原料,经过甲基化、硝基还原、炔基取代、叠氮化以及环化反应合成1-(3-甲氧基苯基)-[4-(4-氟-2-甲氧基-D3苯胺)甲基]-1,2,3-三氮唑。该合成方法简单而且稳定性高,以碘甲烷-D3计,目标产物总收率为60.2%,化学纯度为99.0%,同位素丰度为99.0%。化合物具有广谱抗肿瘤活性,有望成为抗肿瘤药物。
参考文献:
[1] It G R, Singh S B, Hamel E, et al. Isolation and structure of the strong cell growth and tubulin inhibitor combretastain A-4[J]. Experientia,1989, 45(2): 209-211.
[2] Gaukroger K, Hadfield J A, Lawrence N J, et al. Structural requirements for the interaction of combretastatins with tubulin: how important is the trimethoxy unit?[J]. Org Biomol Chem, 2003, 1(17): 3 033-3 037.
[3] Pettit G R, Minardi M D, Rosenberg H J, et al. Antineo plastic agents509:synthesis of fluorcombstat in phosphateandrelated 3-halostilbenes[J]. J Nat Prod, 2005, 68(10): 1 450-1 458.
[4] Pettit G R, Anderson C R, Herald D L, et al .Antineo plastic agents 487.synthesis and biological evaluation of the antineo-plastic agent 3,4-methylene dioxy-5,4′-dimethoxy-3′-amino-Z-stilbene and derived aminoacidamides[J]. J Med Chem, 2003, 46(4): 525-531.
[5] John J H, Madhavi S, Tracy E S, et al. Design, synthesis, biochemical, and biological evaluation of evaluation of nitrogen-containing trifluoro structural modifications of combretastain A-4[J]. Bioorg Med ChemLett, 2008, 18(18): 5 146-5 149.
[6] Maya A B S, Del Rey B, de Clairac R P L, et al. Design, synthesis and cytotoxic activities of naphthylanphthyl analogues of combretastain A-4[J]. Bioorg Med ChemLett, 2000, 10(22) : 2 549-2 551.
[7] Hu L, Li Z R,Wang Y M, et al. Novel pyridinyl and pyrimidinylcarbazole sulfonamides as antiproliferative agents[J]. Bioorg Med Chem Lett, 2007, 17(5): 1 193-1 196.
[8] Ohsumi K, Nakagawa R, Fukuda Y, et al. Novel combretastatinananlogues effective against murine solid tumors: Design and structure-activity relationships[J]. J Med Chem, 1998, 41(16): 3 022-3 032.
[9] Pinney K G, Mejia M P, Villalobos V M, et al. Synthesis and biological evaluation of aryl azide derivatives of combretastatin A-4 as molecular probes for tubulin[J]. Bioorg Med Chem, 2000, 8(10): 2 417-2 425.
[10] Monk K, Siles R, Hadimani M B, et al. Design, synthesis, and biological evaluation of combretastatin nitrogen-containing derivatives as inhibitors of tubulin assembly and vascular disrupting agents[J]. Bioorg Med Chem, 2006, 14(9): 3 231-3 244.
[11] Hatanaka T, Fujita K, Ohsumi K, et al. Novel B -ring modified combretastatin analogues: Syntheses and an tineoplasticactivity[J]. Bioorg Med Chem Lett,1998,8(23): 3 371-3 374.
[12] Rao Y K, Fang S H, Tzeng Y M. Synthesis and biological evaluation of 3′,4′,5′-trimethoxychalcone analogues as inhibitors of nitric oxide production and tumor cell proliferation[J]. Bioorg Med Chem, 2009,17(23) : 7 909-7 914.
[13] 汪忠华. 一种1-取代苯基-4-取代苯胺甲基-1,2,3-三氮唑衍生物及其制备方法和用途:中国,201410834545.2[P].2014-12-23.
[14] 巫辅龙. 二苯基取代氨甲基三氮唑类化合物的合成和活性研究[D]. 上海:上海应用技术大学,2016.
