SAHA联合厄洛替尼对EGFR-TKI耐药肺癌细胞株H1975的生长抑制作用及机制
2018-07-04陈立豪郝吉庆
陈立豪,郝吉庆
肺癌是发病率及死亡率最高的恶性肿瘤,全球每年约有150万人患病,5年生存率不足20%,依据组织学分型,非小细胞肺癌(non-small cell lung cancer, NSCLC) 约占80%[1-2]。随着癌症的个体化治疗及精准医学的提出,基于肿瘤患者的驱动基因突变检测在肿瘤诊断和治疗过程中有着重要的影响。在亚裔、不吸烟、女性、腺癌患者中表皮生长因子(epidermal growth factor receptor,EGFR)基因的突变率较高,约占50%,尤其在我国EGFR突变率为30%,常为外显子19缺失突变(del 19)及外显子21的替换突变(L858R)[3];临床上具有EGFR突变的患者在使用针对EGFR突变的靶向药物如特罗凯(厄洛替尼)、易瑞沙(吉非替尼)等时,早期绝大部分患者都取得不错的临床效果,但在经过治疗一段时间后都会无法避免地出现耐药现象,导致治疗失败[4]。除了基因的改变与肿瘤的发生发展有着重要的关系,表观遗传的改变也起着重要的作用。近年来研究[5]证实,异常的DNA甲基化和组蛋白修饰等表观遗传学的改变,在肿瘤的发展中有着至关重要的作用。组蛋白去乙酰化酶( histone deacetylase,HDAC) 抑制剂通过抑制组蛋白及非组蛋白的去乙酰化,从而抑制细胞增殖,诱导细胞凋亡等来发挥抗肿瘤作用。该研究通过体外单药及联合用药对肺腺癌表皮生子因子酪氨酸激酶抑制剂(epidermal growth factor receptor-tyrosine kinase inhibitor,EGFR-TKI)靶向耐药细胞株H1975的增殖、侵袭影响,探讨HDAC抑制剂伏立诺他 (vorinostat,SAHA)与厄洛替尼联合对肺癌细胞是否具有协同抑制作用以及其可能的作用机制。
1 材料与方法
1.1实验材料人肺腺癌H1975细胞株购自上海中科院细胞库;RPMI-1640购自美国Hyclone公司;胎牛血清(FBS)购自杭州四季青生物科技公司;CCK-8试剂盒购自北京索莱宝科技公司;Transwell小室、Matrigel基质胶购自康宁(中国)科技有限公司;SAHA 及厄洛替尼均购自美国 Selleck 公司;Western blot化学发光剂购自美国Millipore 公司;磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)、丝氨酸/苏氨酸蛋白激酶(the serine-threonine kinase,AKT)、磷酸化丝氨酸/苏氨酸蛋白激酶(phosphorylated serine-threonine kinase,p-AKT)、哺乳动物雷帕霉素靶蛋白(the mammalian target of rapamycin,mTOR)、磷酸化哺乳动物雷帕霉素靶蛋白(phosphorylated mammalian target of rapamycin,p-mTOR)抗体均购自美国Cell Signaling Technology公司。
1.2实验方法
1.2.1细胞培养 H1975细胞细胞培养于含10%胎牛血清、1%青链霉素(100 U/ml)的RPMI-1640培养液中,在 37 ℃、5% CO2饱和湿度培养箱中常规培养,细胞融合80%时进行传代,取对数生长期细胞用于各项实验研究。
1.2.2CCK-8 取对数生长期的细胞,以约5 000细胞每孔接种于96孔板中,培养过夜后待细胞完全贴壁后分为4组作用:① 对照组(不加药);② SAHA组(0.25、0.5、1、2、4、8、16 μmol/L);③ 厄洛替尼组(0.25、0.5、1、2、4、8、16 μmol/L);④ SAHA+厄洛替尼组(SAHA 0.25 μmol/L/L+厄洛替尼 0.25μmol/L, SAHA 0.5 μmol/L+厄洛替尼0.5 μmol/L以此类推),每组设5个复孔,药物作用48 h后,每个孔加入10 μl CCK-8溶液,培养2 h,在酶标仪测450 nm处吸光度(absorbance,A)值,并计算细胞存活率(survival rate,SR),试验重复3次。细胞存活率(%)=(试验组A/对照组A)×100%。根据实验结果,对于后续实验采用SAHA 3 μmol/L,厄洛替尼 19 μmol/L,SAHA( 2 μmol/L)+厄洛替尼(2 μmol/L)。
1.2.3平板克隆实验 将对数生长期细胞,用胰酶消化后吹散成单细胞悬液,取500个每个孔接种于6孔板中,置于37 ℃、5% CO2培养箱中培养24 h,细胞贴壁后,分为4组:① 对照组;② SAHA组;③ 厄洛替尼组;④ SAHA+厄洛替尼联合用药组。每3 d更换一次培养液至细胞集落形成,终止培养。弃培养液,PBS 液洗2次后,纯甲醇固定15 min,结晶紫染色30 min,空气干燥后,显微镜下计数形成的克隆数(≥50个细胞为一个克隆) ,计算各组克隆形成率。克隆形成率(%)=克隆数/接种细胞数×100%。实验重复3次。
1.2.4Transwell小室侵袭实验 将分装好的Matrigel胶(4 ℃)放置过夜使之溶解, 用 4 ℃预冷的无血清培养液稀释至1 mg/ml,取稀释的 Matrigel胶100 μl加入到Transwell小室的上室, 将铺胶后的Transwell小室于 37 ℃温育至少2 h, 以使凝胶形成。胰酶消化4组经过药物作用48 h后细胞,并收集细胞, 用PBS洗涤细胞3次, 用无血清的培养液重悬细胞。取 100 μl的细胞悬液(细胞数约8 000个)加入上室,在 Transwell小室的下室加入含培养液700 μl,37 ℃培养24 h。取出上室, 用棉签擦掉上层细胞, 加入甲醇固定 3 min, 弃固定液, 加入 0.1%结晶紫染液, 染色 20 min, 双蒸水洗3遍, 荧光显微镜下拍照,随机选取3个视野计算,取平均值,实验重复3次。
1.2.5Western blot法 各组细胞处理48 h后收集细胞,加入预冷的RIPA 裂解液( 含PMSF及磷酸化酶抑制剂) 将细胞重悬,于冰上裂解30 min后,4 ℃、12 000 r/min离心15 min,吸取上清液行Bradford 法蛋白浓度测定,然后行SDS-PAGE 电泳,Western blot法转移蛋白,一抗4 ℃过夜,二抗37 ℃孵育2 h,应用ECL化学发光试剂盒进行显影,电泳条带用用扫描仪采集X线片图像,使用Quantity One 软件,以β-actin为内参照,对各目的条带进行密度分析。

2 结果
2.1SAHA、Erlotinib单药及联合用药对H1975细胞存活率的影响CCK8结果显示,药物作用48 h后,SAHA组(1、2、4、8、16 μmol/L)较对照组细胞抑制率差异有统计学意义(t=7.794、16.429、42.212、209.000、143.760,P<0.05);Erlotinib组(1、2、4、8、16 μmol/L)较对照组细胞抑制率差异有统计学意义(t=6.379、8.220、15.057、18.187、42.500,P<0.05);联合用药组(1、2、4、8、16 μmol/L)较对照组细胞抑制率差异有统计学意义(t=79.674、67.278、75.215、 71.279、108.098,P<0.05);可见随药物浓度增加,药物对细胞的抑制作用也逐渐增强,呈现浓度依赖性。SAHA和Erlotinib对H1975细胞的半数抑制浓度(half maximal inhibitory concentration,IC50)值分别为(3.16±0.33)和(19.31±2.53)μmol/L,而联合用药组IC50值为(2.11±0.25)μmol/L,差异有统计学意义(F=127.299,P<0.05),见图1。不同浓度的SAHA联合Erlotinib抑制剂处理H1975细胞48 h后,两药联合后增殖抑制作用明显增强,各联合药物剂量组的联合指数(combination index,CI)均<1,表现出协同效应(图2)。
2.2SAHA、Erlotinib单药及联合用药对H1975肿瘤细胞的克隆形成SAHA组对细胞克隆形成率为41.90%,Erlotinib组形成率48.11%,而联合用药组17.89%。结果显示联合用药组较单药组能够显著抑制肿瘤细胞的克隆形成,差异有统计学意义(F=29.350,P<0.001),见图3。
2.3SAHA、厄洛替尼单药及联合用药对H1975细胞侵袭能力的影响实验结果显示:SAHA组及Erlotinib组细胞侵袭能力较对照组降低,差异有统计学意义(t=6.072、3.838,P<0.05);两药联合组与单药组细胞相比,细胞侵袭到Transwell 小室Matrigel 胶下面的细胞数显著降低,差异有统计学意义(F=29.350,P<0.05),见图4。
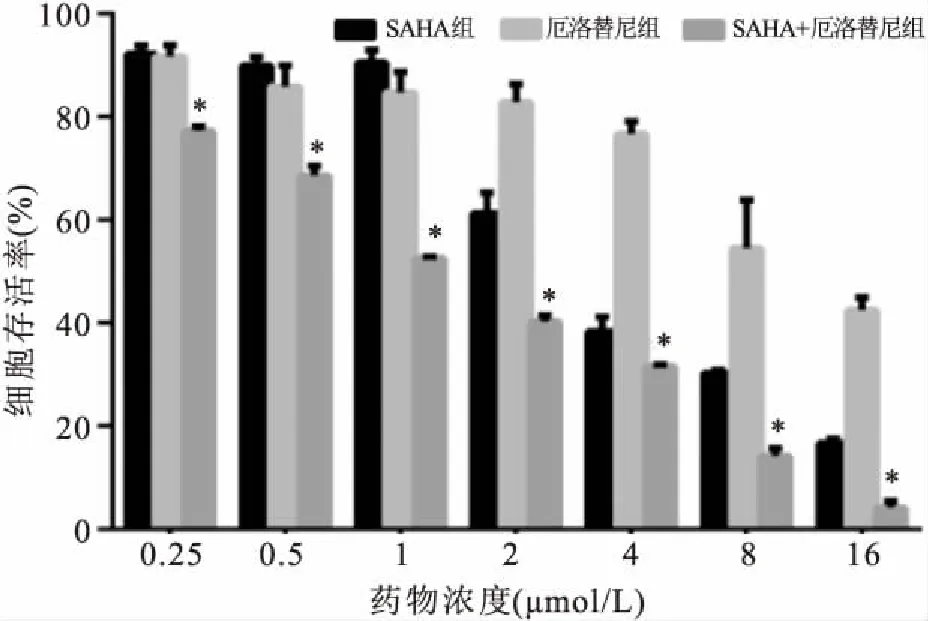
图1 SAHA、厄洛替尼单药及联合用药对H1975细胞的增殖活力作用
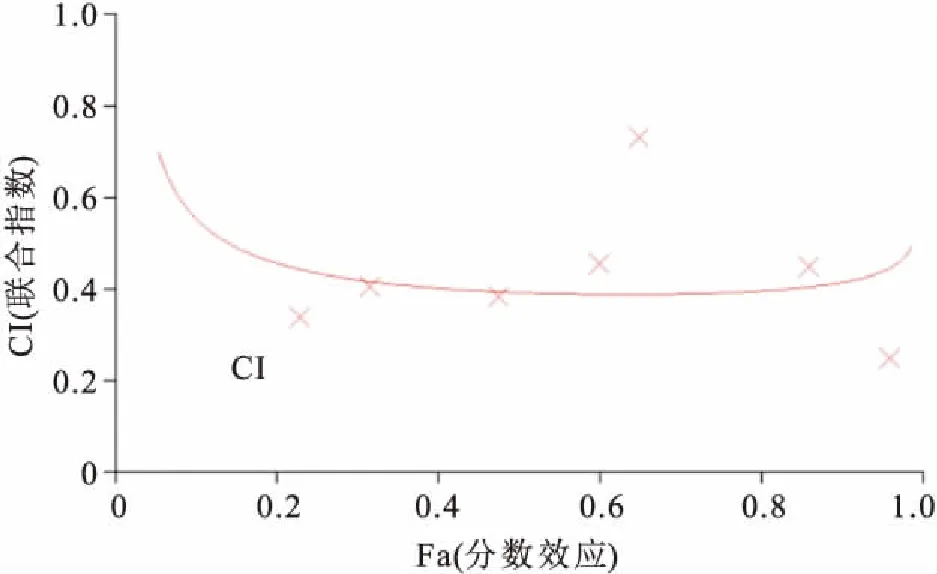
图2 SAHA联合Erlotinib 对H1975细胞的联合作用效应
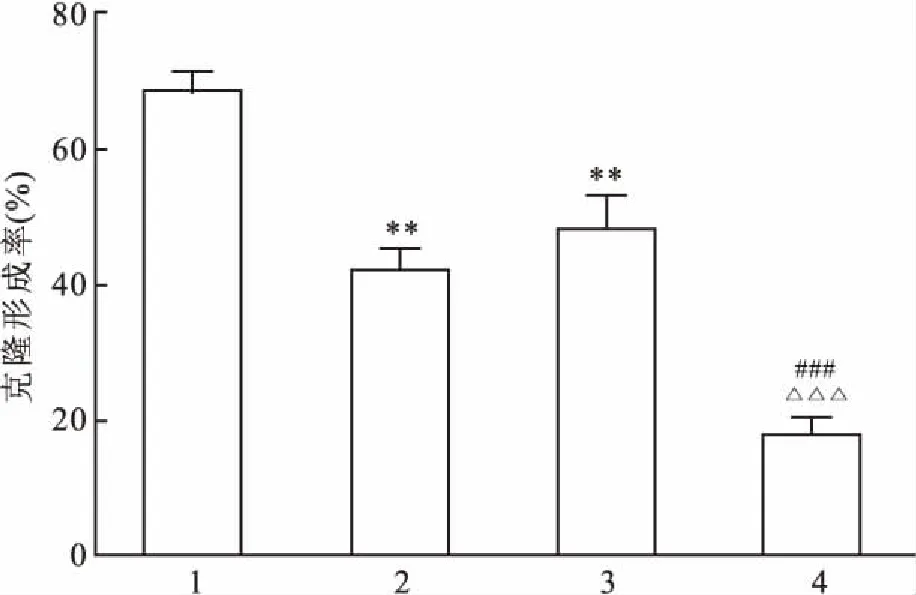
图3 SAHA、Erlotinib单药及联合用药对H1975肿瘤细胞的克隆率形成
1:对照组;2:SAHA组;3:厄洛替尼组;4:SAHA+厄洛替尼组;与对照组比较:**P<0.01;与SAHA组比较:###P<0.001;与厄洛替尼组比较:△△△P<0.001
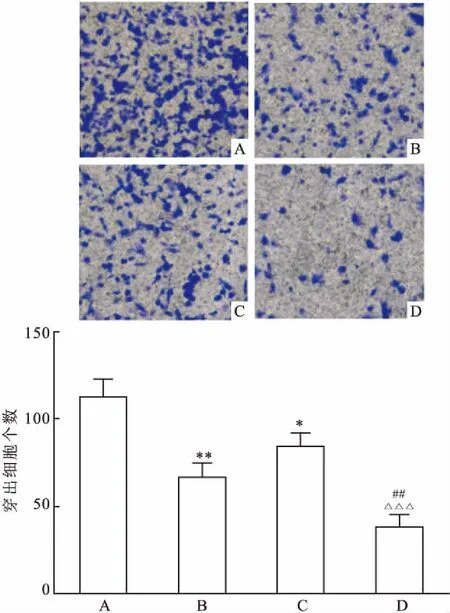
图4 Transwell 小室培养48 h后各组迁移到下室的细胞结晶紫染色图 ×200
A:对照组;B:SAHA组;C:厄洛替尼组;D:SAHA+厄洛替尼组;与对照组比较:*P<0.05,**P<0.01;与SAHA组比较:##P<0.001;与厄洛替尼组比较:△△△P<0.001
2.4SAHA、Erlotinib单药及联合用药对H1975细胞内PI3K/AKT/mTOR信号通路影响H1975细胞经过药物处理48 h后,应用Western blot检测研究结果显示:PI3K蛋白表达在各处理组都有明显降低;p-AKT蛋白表达量在SAHA组降低,而Erlotinib组无明显改变;p-mTOR蛋白表达量在各组都降低。两药联合组较其余各组PI3K、p-AKT、p-mTOR蛋白表达量均明显减低,差异具有统计学意义(F=84.198、349.523、119.197,P<0.01),见图5。
3 讨论
肺癌由于早期症状不典型,大部分患者在诊断时已处于中晚期,失去手术机会。化疗和靶向治疗是晚期NSCLC患者的主要治疗手段。相对于传统化疗,靶向药物具有副反应轻、选择性较高、耐受性好、服用方便、依从性高等特点[6]。EGFR是原癌基因HER1表达产物,为一跨细胞膜糖蛋白,属于酪氨酸激酶受体,在多种肿瘤中均可发现EGFR的高表达或异常表达。EGFR-TKI如厄洛替尼等,通过与EGFR结构域中的ATP结合位点相结合,阻断EGFR下游信号通路的传导,达到抑制肿瘤细胞增殖作用;还可以抑制丝裂原活化蛋白激酶的活化,促进肿瘤细胞凋亡以及抑制肿瘤细胞的血管形成。对于EGFR突变阳性患者有明显疗效,使患者无进展生存期显著提高。但绝大部分患者都不可避免的在一年内出现耐药,导致病情恶化。常见的耐药机制是T790M突变(50%)、MET基因扩增(20%)等[7],尽管临床上对于一代靶向药物的耐药而开发出二代、三代靶向药物,但是大部分患者使用一段时间后效果并不是非常理想,故对于这些EGFR-TKI治疗失败的患者该如何治疗是临床医师比较棘手的问题。
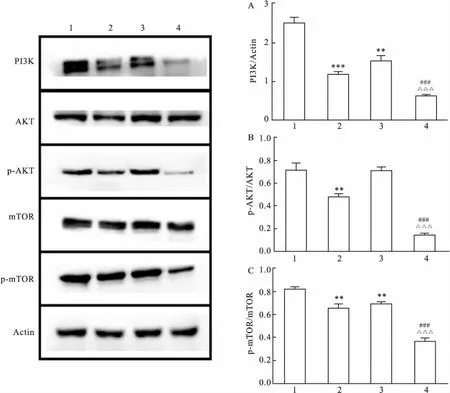
图5 药物对H1975细胞PI3K/AKT/mTOR信号通路的影响
1:对照组;2:SAHA组;3:厄洛替尼组;4:SAHA+厄洛替尼组;与对照组比较:**P<0.01,***P<0.001;与SAHA组比较:###P<0.001;与厄洛替尼组比较:△△△P<0.001
表观遗传是指基因表达或蛋白质的表达改变不涉及碱基序列的变化,但可以稳定遗传的现象。越来越多的研究[8]表明,随着年龄增长、外界环境刺激等因素,细胞的正常表观遗传状态会被打破,导致癌基因的异常激活或抑癌基因的失活,导致肿瘤的发生、发展。表观遗传修饰主要包括DNA甲基化、组蛋白修饰、非编码RNA的调控、染色质重塑等[9]。近年来随着对表观遗传学研究,组蛋白的异常修饰调节与肿瘤发生有密切关系;正常情况下组蛋白处于乙酰化及去乙酰化的动态平衡之中,乙酰化受组蛋白乙酰基转移酶调控;去乙酰化则受HDAC的调控。在肿瘤细胞中,HDAC活性明显增强,平衡状态打破,使得抑癌基因表达下调的同时致癌基因表达上调[10]。
HDAC抑制剂作为一种高效低毒的靶向抗肿瘤药物正日益引起人们的关注。 它可以通过抑制HDACs,调节组蛋白和非组蛋白的乙酰化水平 ,从而达到调控基因表达的目的,以逆转变异细胞的表型、抑制细胞的增殖、诱导肿瘤细胞周期阻滞、分化、通过内源性和外源性途径诱导细胞凋亡、抑制血管生成和肿瘤侵袭等[11]。SAHA是一种广谱的组蛋白抑制剂,于2006年被FDA批准上市,用于病情恶化或病情反复者的皮肤T细胞淋巴瘤的治疗。当其他药物治疗效果不佳时,该药表现出良好的耐受性[12]。研究[13]表明,SAHA和卡铂、紫杉醇还可联合治疗晚期非小细胞肺癌,并且SAHA在多项肿瘤的联合化疗中均显示出其协同或增强作用。
本研究选用具有T790M突变的肺癌细胞株H1975,探讨两药体外联合应用对肺癌细胞的影响及作用机制,结果显示SAHA与厄洛替尼联合用药具有协同作用,同单独用药组比较能够显著抑制H1975肿瘤细胞的生长增殖、克隆及侵袭能力;同时由于PI3K/AKT/mTOR信号传导通路的过渡激活与肿瘤的增殖、侵袭转移、抗凋亡有着重要的关系;本研究发现联合用药组对PI3K/AKT/mTOR信号通路有着显著的抑制作用。
综上所述,厄洛替尼联合HDAC抑制剂SAHA对具有T790M突变的H1975细胞株具有良好的协同作用,可增强细胞对厄洛替尼的敏感性,为临床上对于EGFR-TKI 获得性耐药患者的治疗提供新的实验室依据及治疗策略。但本实验仅为初步的体外细胞研究,尚需进一步的动物实验及临床试验来验证其在体内的有效性及安全性。
[1] Jacek P, Beata J P, Joanna R, et al. Quality of life of patients with lung cancer[J]. Onco Targets Ther, 2016, 9(1):1023-8.
[2] Siegel R L, Miller K D,Jemal A.Cancer statistics[J].CA Cancer J Clin,2015, 65(1):5-29.
[3] Shi Y, Au S K, Thongprasert S, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER)[J]. J Thorac Oncol, 2014, 9(2):154-62.
[4] Sudo M, Tan M C, Mori S, et al. Inhibiting proliferation of gefitinib-resistant, non-small cell lung cancer[J]. Cancer Chemother Pharmacol, 2013,71(5):1325-34.
[5] Sarkar S, Horn G, Moulton K, et al. Cancer development, progression, and therapy: an epigenetic overview[J]. Int J Mol Sci, 2013,14(10):21087-113.
[6] 权修权, 朴惠顺, 康 琳,等. 抗肿瘤靶向药物研究现状[J]. 中国药理学通报, 2015,19(5):610-4.
[7] 刘慧慧,王孟昭,胡 克,等.EGFR-TKI在非小细胞肺癌中耐药机制的研究进展[J].中国肺癌杂志, 2013,16(10):535-40.
[8] 侯海金, 张振华, 闫慧明. 表观遗传学在肿瘤中的作用[J]. 中华临床医师杂志(电子版), 2016,10(18):2780-4.
[9] Li J, Hao D, Li W, et al. Epigenetic targeting drugs potentiate chemotherapeutic effects in solid tumor therapy[J]. Sci Rep, 2017, 7(1):4035-47.
[10] Takashina T, Kinoshita I, Kikuchi J, et al. Combined inhibition of EZH2 and histone deacetylases as a potential epigenetic therapy for non-small-cell lung cancer cells[J]. Cancer Sci, 2016, 107(7):955-62.
[11] Wang L, Xiang S, Williams K A, etal. Depletion of HDAC6 enhances cisplatin-induced DNA damage and apoptosis in non-small cell lung cancer cells[J]. PLoS One, 2012, 7(9):e44265-e44276.
[12] Mann B S, Johnson J R, Cohen M H, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma.[J].Oncologist, 2007, 12(10):1247-52.
[13] 姚毅武, 姚和权, 蒋 晟,等. 组蛋白去乙酰化酶抑制剂抗肿瘤临床研究进展[J]. 中国新药杂志, 2013,22(3):294-300.
