HIPK2基因对缺氧复氧诱导的NRK-52E肾小管上皮细胞活力和凋亡及JAK2/STAT3信号通路的影响*
2018-06-30朱彬蔚卞蓉蓉陈冬平梅长林
朱彬蔚,卞蓉蓉,陈冬平,梅长林
(第二军医大学长征医院肾内科, 上海 200001)
急性肾损伤是临床上常见的一种综合征,缺血再灌注损伤(ischemia reperfusion injury,IRI)是引起该病的一个重要因素[1]。近些年来,在临床上常采用导管术、溶栓疗法和肾脏移植等新技术重新恢复缺血肾组织血流,但却引起肾脏IRI的发生[2]。肾小管上皮细胞(renal tubular epithelial cells,RTEC)对脓毒血症、中毒和缺血缺氧等损伤因素较敏感,缺氧损伤是引起各种肾脏疾病发生的重要原因,主要的病变是小管上皮细胞的凋亡和坏死,而小管上皮细胞的增殖、去分化及再分化是损伤后修复的基础[3]。目前已发现在肾脏缺氧损伤后有多种热点分子,如血管内皮生长因子、结缔组织生长因子和细胞间黏附因子等参与了修复过程,但却不是有效的治疗手段[4]。因此,寻找有效的急性肾损伤治疗靶点具有重要意义。同源结构域相互作用蛋白激酶2(homeo-domain-interacting protein kinase 2,HIPK2)是HIPK家族中的一员,是一种丝/苏氨酸蛋白激酶,参与调控基因表达,受到信号诱导后激活,在细胞增殖和凋亡等信号转导过程中有重要作用[5]。近些年的研究表明,HIPK2是一个肿瘤抑制基因,可影响肝癌和结直肠癌等多种肿瘤的生物学特性[6-7];近期研究报道,HIPK2是肾脏纤维化过程中的一个关键调节因子,其高度活跃时可引起肾脏纤维化的发生[8]。而HIPK2在急性肾损伤中的作用还未清楚。因此,本研究通过将靶向HIPK2的小干扰RNA(small interfering RNA,siRNA)转染大鼠肾近曲小管上皮细胞株NRK-52E,并进行缺氧复氧(hypoxia and reoxyge-nation,H/R)处理,检测细胞增殖和凋亡情况,并研究其作用机制。
材 料 和 方 法
1 实验材料
大鼠肾近曲小管上皮细胞株NRK-52E购自ATCC;胎牛血清和胰蛋白酶均购自Gibco; DMEM培养基购自HyClone; LipofectamineTM2000转染试剂盒购自Invitrogen; CCK-8试剂盒购自碧云天生物技术研究所;Annexin V-FITC/碘化丙啶(propidium iodide,PI)细胞凋亡试剂盒购自南京凯基生物技术公司;二喹啉甲酸(bicinchoninic acid,BCA)试剂盒购自Nerck Millipore;Fluo-3/AM购自Biotium;兔抗人cleaved Caspase-3和caspase-12多抗、鼠抗人Bcl-2单抗、鼠抗人Bax单抗、兔抗人磷酸化Janus激酶2(phosphorylated Janus kinase 2,p-JAK2)单抗和兔抗人磷酸化信号转导及转录激活因子3(phosphorylated signal transducer and activator of transcription 3,p-STAT3)单抗均购自Abcam;酶标仪购自Bio-Rad;流式细胞仪购自BD。
2 实验方法
2.1细胞培养及分组 取出冻存在液氮罐中的NRK-52E细胞,37 ℃水浴快速溶解,溶解后细胞置于37 ℃、5% CO2及95% O2的培养箱中用含10%胎牛血清的DMEM培养液培养,换液去除未贴壁细胞,细胞生长融合至80%以上,用胰蛋白消化后进行传代。细胞达80%融合后换成不含胎牛血清的高糖培养液同步化培养24 h,随机分为正常对照组(control组)、阴性对照组(HIPK2-NC组)、H/R组和HIPK2-siRNA+H/R组。
2.2HIPK2-siRNA转染NRK-52E细胞效率的检测 HIPK2-siRNA转染NRK-52E细胞参照LipofectamineTM2000说明书。取转染48 h的细胞,加入细胞裂解液提取细胞中蛋白,BCA试剂盒定量蛋白,制备5%的浓缩胶和12%的分离胶,取40 μg蛋白样品与上样缓冲液充分混匀,100 ℃煮沸变性5 min,行SDS-PAGE分离(浓缩胶电压为80~100V,电泳约30 min;分离胶电压120 V,电泳约1.5 h),电泳结束后转PVDF膜,5%脱脂奶粉封闭, I 抗孵育(HIPK2和内参照GAPDH皆按照1∶1 000稀释),4 ℃孵育过夜,洗膜,加入HRP标记的羊抗兔抗体 II 抗(1∶2 000稀释),洗膜,加入配置好的化学发光试剂,室温孵育2 min,曝光、显影、定影。Quantity One软件对蛋白灰度值进行分析。
2.3缺氧复氧细胞模型的制备 取转染48 h后的细胞,缺氧制备前将培养液换成磷酸盐缓冲液溶液,置于含有体积分数为1% O2、4% CO2及95% N2混合气体缺氧的37 ℃培养箱中培养,1 h后,换为正常培养基培养,置于5% CO2及95% O2的培养箱内2 h,收集细胞。
2.4CCK-8法检测细胞活力 各组细胞活力采用CCK-8法检测。每孔加入CCK-8液10 μL,37 ℃孵育4 h,酶标仪以波长490 nm测定各孔的吸光度(A)值。实验重复3次。以吸光度值反映细胞活力。
2.5流式细胞术检测细胞凋亡 采用Annexin V-FITC/PI双染法检测各组细胞凋亡情况。将5×105个NRK-52E细胞接种于6孔板中,按照前面分组处理后,胰蛋白酶消化收集细胞,离心,弃掉上清,预冷的磷酸盐缓冲液洗涤细胞,离心,弃上清,加入结合缓冲液500 μL重悬细胞,加入Annexin V-FITC 5 μL,充分混匀后,置于4 ℃避光环境中孵育15 min,再加入PI 5 μL,置于室温避光环境中5~15 min,1 h内上流式细胞仪检测各组细胞的凋亡情况。实验重复3次。
2.6Ca2+荧光强度检测 采用流式细胞仪检测游离的Ca2+荧光强度。胰蛋白酶消化后收集细胞,离心,磷酸盐缓冲液洗涤2遍,细胞中加入1 μmol/L的钙敏感荧光探针Fluo-3/AM,充分振荡混合均匀,置于37 ℃避光条件下孵育45 min,磷酸盐缓冲液洗涤除去没有结合的燃料,继续15 min孵育后送检(1 000个细胞)。Fluo-3/AM的发射波长及激发波长分别为526 nm和488 nm。细胞内游离Ca2+浓度通过荧光强度反映。实验重复3次。
2.7Western blotting检测蛋白水平 细胞内增殖相关蛋白Ki67、凋亡相关蛋白cleaved caspase-3、 caspase-12、Bcl-2、Bax及JAK2/STAT3信号通路p-JAK2和p-STAT3蛋白水平的检测参照方法2.2进行。
3 统计学处理
所有实验数据采用SPSS 21.0软件进行分析,计量资料用均数±标准差(mean±SD)表示,多组差异比较采用单因素方差分析,组间两两比较采用Bonferroni校正的t检验,以P<0.05为差异有统计学意义。
结 果
1 HIPK2-siRNA转染NRK-52E细胞效果
HIPK2-siRNA组HIPK2的蛋白表达显著低于control组(P<0.05),而在HIPK2-NC组HIPK2的蛋白表达与control组差异无统计学显著性,见图1。这说明转染HIPK2-siRNA可显著降低HIPK2的表达。
2 HIPK2-siRNA转染可促进H/R诱导的NRK-52E细胞活力
H/R组的细胞活力显著低于control组(P<0.05),而HIPK2-siRNA+H/R组细胞活力显著高于H/R组(P<0.05),见图2。
3 HIPK2-siRNA转染可抑制H/R诱导的NRK-52E细胞凋亡
H/R组细胞凋亡率显著高于control组(P<0.05),而HIPK2-siRNA+H/R组细胞凋亡率显著低于H/R组(P<0.05),见图3。
4 HIPK2-siRNA转染可降低H/R诱导的NRK-52E细胞的Ca2+荧光强度
H/R组Ca2+荧光强度值显著高于control组(P<0.05),而HIPK2-siRNA+H/R组Ca2+荧光强度值显著低于H/R组(P<0.05),见图4。
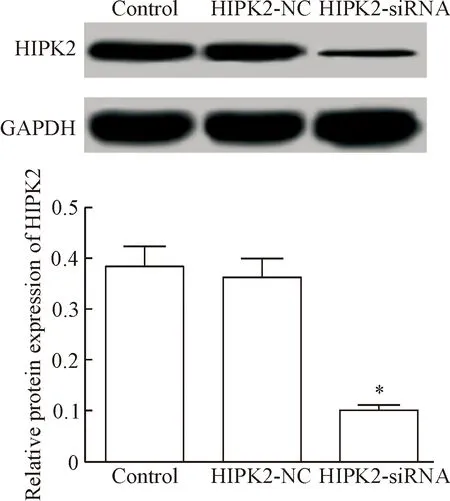
Figure 1. The protein expression of HIPK2 after HIPK2-siRNA was transfected into NRK-52E cells. Mean±SD.n=3.*P<0.05vscontrol group.
图1HIPK2-siRNA转染NRK-52E细胞后HIPK2的蛋白表达

Figure 2. The effect of HIPK2-siRNA transfection on the viability of NRK-52E cells induced by H/R. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsH/R group.
图2HIPK2-siRNA转染对H/R诱导的NRK-52E细胞活力的影响
5 HIPK2-siRNA转染对H/R诱导的NRK-52E细胞增殖及凋亡相关蛋白表达的影响
H/R组的Ki67和Bcl-2蛋白表达显著低于control组(P<0.05),cleaved caspase-3、 caspase-12和Bax的蛋白表达显著高于control组(P<0.05);而HIPK2-siRNA+H/R组Ki67和Bcl-2蛋白表达显著高于H/R组(P<0.05),cleaved caspase-3、caspase-12和Bax的蛋白表达显著低于H/R组(P<0.05),见图5。
6 HIPK2-siRNA转染可下调H/R诱导的NRK-52E细胞JAK2/STAT3信号通路蛋白的磷酸化水平
H/R组的p-JAK2和p-STAT3蛋白表达均显著高于control组(P<0.05),而HIPK2-siRNA+H/R组的p-JAK2和p-STAT3蛋白水平显著低于H/R组(P<0.05),见图6。

Figure 3. The effect of HIPK2-siRNA transfection on the apoptosis of NRK-52E cells induced by H/R. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsH/R group.
图3HIPK2-siRNA转染对H/R诱导的NRK-52E细胞凋亡的影响
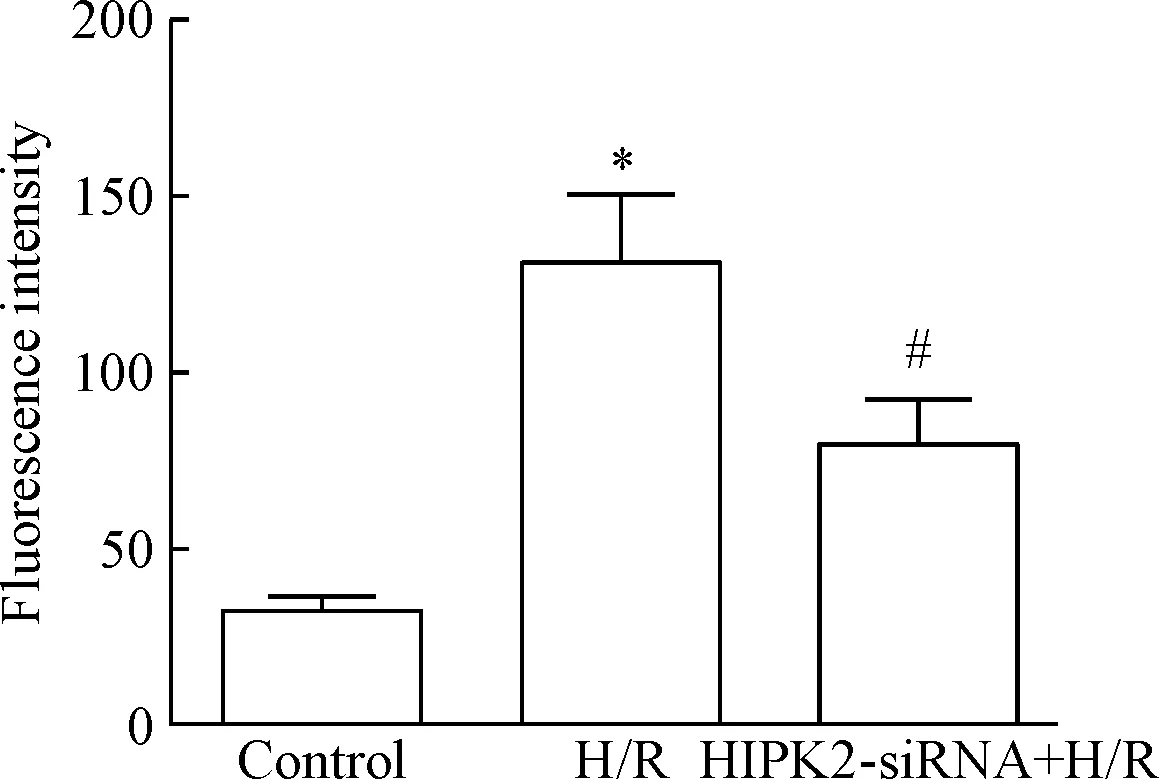
Figure 4. The effect of HIPK2-siRNA transfection on Ca2+fluorescence intensity of NRK-52E cells induced by H/R. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsH/R group.
图4HIPK2-siRNA转染对H/R诱导的NRK-52E细胞Ca2+荧光强度的影响
讨 论
肾IRI是引起缺血性肾衰竭的一个主要原因,如何有效防治肾IRI是国内外学者关注的热点[9]。肾IRI的发生机制较为复杂,RTEC凋亡是其发生的一个重要机制,主要与钙超载、氧自由基增多和能量代谢障碍等因素有密切联系[10],因此研究有效的肾脏损伤治疗方法尤为重要。HIPKs家族分为HIPK1、HIPK2和HIPK3。小鼠HIPK2基因定位于6B染色体上,人类HIPK2基因定位于7q32~34,是一种细胞核内的丝苏氨酸蛋白激酶。近些年的研究发现,HIPK2是一个有多种功能潜在的肿瘤抑制因子,可通过调控肿瘤细胞中一些关键分子通路,而抑制肿瘤生长及使药物敏感性增强[11-12]。随着对HIPK2研究的深入,其在肾脏中的作用受到广泛关注。研究发现,HIPK2可通过调节Notch、p53和Smad3等通路在肾脏中发挥促炎和纤维化作用[13-14]。最新研究发现,HIPK2表达的抑制可减缓肾脏纤维化的进程,可作为肾脏纤维化治疗的靶点[15]。但关于HIPK2在肾损伤中的研究不多。
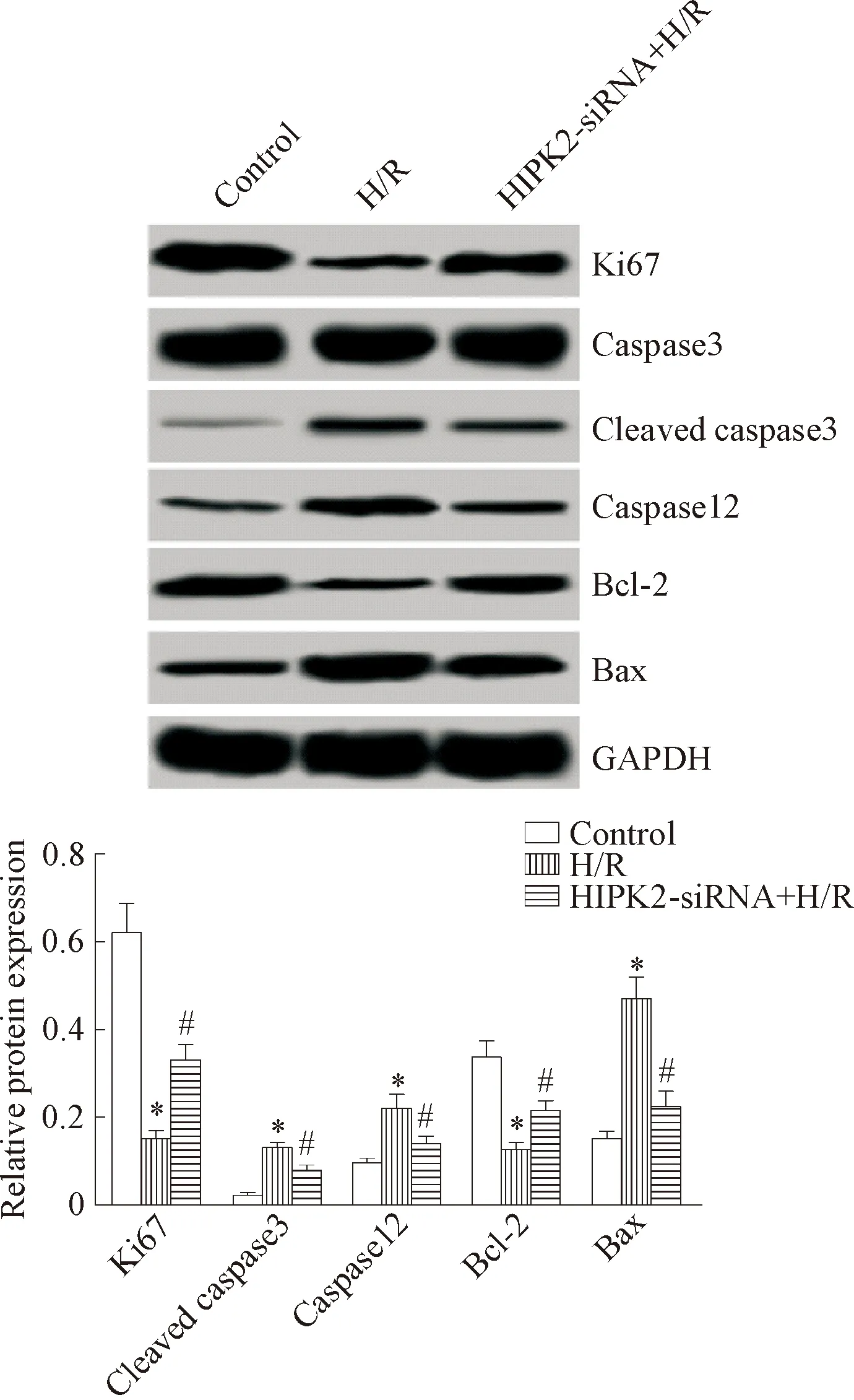
Figure 5. The effect of HIPK2-siRNA transfection on the expression of proliferation-and apoptosis-related proteins in the NRK-52E cells induced by H/R. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsH/R group.
图5HIPK2-siRNA转染对H/R诱导的NRK-52E细胞增殖及凋亡蛋白表达的影响
本研究将靶向HIPK2的siRNA转染NRK-52E细胞,细胞中HIPK2的表达明显降低,说明转染后可明显抑制HIPK2表达。细胞经过H/R处理,通过CCK-8法及流式细胞术分别检测细胞活力及凋亡情况,发现H/R处理细胞活力明显降低,凋亡率增加,而HIPK2-siRNA+H/R处理可改善这一现象。这提示沉默HIPK2对急性肾损伤的肾小管起到保护作用。Ki67是一种与增殖细胞相关的核抗原,是检测细胞增殖活性比较准确、理想、可靠的抗原,已在多种疾病的研究和探讨中得到广泛的应用[16-18]。细胞凋亡信号有线粒体和死亡受体途径2条经典的传导通路。近些年的研究发现,过度的内质网应激(endoplasmic reticulum stress,ERS)可引起细胞凋亡启动,是一条新的引起细胞凋亡的信号传导通路,可特异性使caspase-12激活,而caspase-12可裂解caspase-3等一些下游效应蛋白酶,最终引起细胞凋亡[19-20]。研究发现,H/R刺激肾上皮细胞可触发ERS,引起caspase-12的激活[21]。Ca2+可参与多种细胞功能的调节,是细胞内正常生理功能维持的重要物质基础,研究发现,I/R损伤可增加细胞内Ca2+,活化caspase-12,从而诱导细胞凋亡[22]。Bcl-2和Bax是Bcl-2家族的2个重要蛋白,Bcl-2/Bax的比例是引起细胞凋亡启动的关键因素,Bcl-2/Bax比率上调可使细胞凋亡抑制,Bcl-2/Bax比率下调可导致细胞凋亡增多[23]。研究发现,在肾脏疾病中Bcl-2和Bax发挥至关重要的作用[24]。本实验结果显示,沉默HIPK2促进H/R处理的NRK-52E细胞增殖的机制是上调Ki67,抑制凋亡的机制是上调Bcl-2表达,下调caspase-12、cleaved caspase-3和Bax表达。

Figure 6. The effect of HIPK2-siRNA transfection on JAK2/STAT3 signaling in NRK-52E cells induced by H/R. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsH/R group.
图6HIPK2-siRNA转染对H/R诱导的NRK-52E细胞JAK2/STAT3信号的影响
JAK2/STAT3信号通路参与细胞的增殖、凋亡、分化、免疫调节和迁移等生理过程,正常RTEC有低水平活化,主要介导修复和增生[25-26]。近些年在肾脏疾病中的调控作用受到广泛关注,已被证实其与足细胞、肾小管上皮细胞、系膜细胞等有密切关系[27]。JAK2/STAT3信号通路的激活可引起RTEC凋亡,可能参与乙肝病毒对肾组织直接损伤的致病机制[28];抑制JAK2/STAT3信号可降低RTEC的转分化[29];H/R条件可激活JAK2/STAT3,而抑制其活性后对RTEC起保护作用[30]。本研究结果显示,HIPK2表达的抑制可下调NRK-52E细胞JAK2/STAT3信号表达,这与前人在肾脏中的作用研究是一致的。
综上所述,抑制HIPK2基因可促进缺氧复氧诱导的NRK-52E肾小管上皮细胞生长,下调JAK2/STAT3信号通路,降低凋亡率,上调Ki67,从而上调Bcl-2表达,同时下调caspase-12、caspase-3和Bax表达。HIPK2基因可能是急性肾损伤治疗的一个新的靶点。但本研究还未进行体内实验研究,存在一定的局限性,这也是我们后期实验研究的重点。
[参 考 文 献]
[1] Kim MG, Cho EJ, Lee JW, et al. The heat-shock protein-70-induced renoprotective effect is partially mediated by CD4+CD25+Foxp3+regulatory T cells in ischemia/reperfusion-induced acute kidney injury[J]. Kidney Int, 2014, 85(1):62-71.
[2] Chawla LS, Eggers PW, Star RA, et al. Acute kidney injury and chronic kidney disease as interconnected syndromes[J]. N Engl J Med, 2014, 371(1):58-66.
[3] Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development[J]. Nat Med, 2015, 21(1):37-46.
[4] 武予霞, 朱海云, 李银平. 低氧诱导因子在肾脏疾病中作用的研究进展[J]. 中国中西医结合急救杂志, 2016, 23(2):222-224.
[5] Hashimoto K, Tsuji Y. Arsenic-induced activation of the homeodomain-interacting protein kinase 2 (HIPK2) to cAMP-response element binding protein (CREB) axis[J]. J Mol Biol, 2017, 429(1):64-78.
[6] Xu J, Zhou J, Li MS, et al. Transcriptional regulation of the tumor suppressor FHL2 by p53 in human kidney and liver cells[J]. PLoS One, 2014, 9(8): e99359.
[7] 郑 荟, 魏光伟. HIPK2 抑制非小细胞肺癌上皮间质转化及迁移侵袭的作用及机制[J]. 山东大学学报: 医学版, 2016, 54(11):7-12.
[8] Fan Y, Wang N, Chuang P, et al. Role of HIPK2 in kidney fibrosis[J]. Kidney Int Suppl, 2014, 4(1):97-101.
[9] 郝 斌, 王 切, 王素玲, 等. EC-SOD 在肾缺血再灌注损伤大鼠脑内的表达变化[J]. 河北医科大学学报, 2017, 38(5):552-556.
[10] Yang Y, Zhang ZX, Lian D, et al. IL-37 inhibits IL-18-induced tubular epithelial cell expression of pro-inflammatory cytokines and renal ischemia-reperfusion injury[J]. Kidney Int, 2015, 87(2):396-408.
[11] Lee S, Shang Y, Redmond SA, et al. Activation of HIPK2 promotes ER stress-mediated neurodegeneration in amyotrophic lateral sclerosis[J]. Neuron, 2016, 91(1):41-55.
[12] Lin J, Zhang Q, Lu Y, et al. Downregulation of HIPK2 increases resistance of bladder cancer cell to cisplatin by regulating Wip1[J]. PLoS One, 2014, 9(5):e98418.
[13] Huang Y, Tong J, He F, et al. miR-141 regulates TGF-β1-induced epithelial-mesenchymal transition through repression of HIPK2 expression in renal tubular epithelial cells[J]. Int J Mol Med, 2015, 35(2):311-318.
[14] Xiong C, Masucci MV, Zhou X, et al. Pharmacological targeting of BET proteins inhibits renal fibroblast activation and alleviates renal fibrosis[J]. Oncotarget, 2016, 7(43):69291-69308.
[15] Chuang PY, Menon MC, He JC. Molecular targets for treatment of kidney fibrosis[J]. J Mol Med, 2013, 91(5):549-559.
[16] 郑 超, 王 卓, 曹余光. 前列腺腺癌组织中果蝇Zeste基因增强因子2、磷酸酶与张力蛋白同源蛋白表达及对Ki67增殖指数的影响[J]. 中国老年学杂志, 2016, 36(12):2946-2947.
[17] Ellis MJ, Suman VJ, Hoog J, et al. Ki67 proliferation index as a tool for chemotherapy decisions during and after neoadjuvant aromatase inhibitor treatment of breast cancer: results from the American college of surgeons oncology group Z1031 trial (Alliance)[J]. J Clin Oncol, 2017, 35(10): 1061-1069.
[18] 刘 岸, 黄成钢, 徐 佳. 慢病毒介导的 GHSR1a shRNA 对结直肠癌细胞生长的影响[J]. 中国病理生理杂志, 2016, 32(7): 1227-1234..
[19] Huang Y, Li X, Wang Y, et al. Endoplasmic reticulum stress-induced hepatic stellate cell apoptosis through calcium-mediated JNK/P38 MAPK and calpain/caspase-12 pathways[J]. Mol Cell Biochem, 2014, 394(1-2):1-12.
[20] Goswami P, Gupta S, Biswas J, et al. Endoplasmic reti-culum stress plays a key role in rotenone-induced apoptotic death of neurons[J]. Mol Neurobiol, 2016, 53(1):285-298.
[21] Fan T, Chen L, Huang Z, et al. Autophagy activation by rapamycin before hypoxia-reoxygenation reduces endoplasmic reticulum stress in alveolar epithelial cells[J]. Cell Physiol Biochem, 2017, 41(1):79-90.
[22] Wang Y, Wei S, Wang YL, et al. Protective effects of circulating microvesicles derived from myocardial ischemic rats on apoptosis of cardiomyocytes in myocardial ische-mia/reperfusion injury[J]. Oncotarget, 2017, 8(33):54572-54582.
[23] 陆志伟, 程玉生, 赵春阳, 等. Shp2 在肺腺癌细胞 A549 中的抑癌作用及机制研究[J]. 中国病理生理杂志, 2016, 32(9):1589-1593.
[24] Shen S, Zhou J, Meng S, et al. The protective effects of ischemic preconditioning on rats with renal ischemia-reperfusion injury and the effects on the expression of Bcl-2 and Bax[J]. Exp Ther Med, 2017, 14(5):4077-4082.
[25] Miao L, Liu K, Xie M, et al. miR-375 inhibitsHelicobacterpylori-induced gastric carcinogenesis by blocking JAK2-STAT3 signaling[J]. Cancer Immunol Immunother, 2014, 63(7):699-711.
[26] 李敏利, 许小兵, 王 彬, 等. 促炎因子在实验性重症急性胰腺炎早期对 JAK2/STAT3 信号通路的影响[J]. 安徽医科大学学报, 2014, 49(10):1392-1395.
[27] Yu J, Wu H, Liu ZY, et al. Advanced glycation end products induce the apoptosis of and inflammation in mouse podocytes through CXCL9-mediated JAK2/STAT3 pathway activation[J]. Int J Mol Med, 2017, 40(4):1185-1193.
[28] He P, Zhang D, Li H, et al. Hepatitis B virus X protein modulates apoptosis in human renal proximal tubular epithelial cells by activating the JAK2/STAT3 signaling pathway[J]. Int J Mol Med, 2013, 31(5):1017-1029.
[29] Wang HY, Zhang C, Xiao QF, et al. Hepatocyte growth factor inhibits tubular epithelial-myofibroblast transdiffe-rentiation by suppression of angiotensin II via the JAK2/STAT3 signaling pathway[J]. Mol Med Rep, 2017, 15(5):2737-2743.
[30] Lv J, Wang X, Liu SY, et al. Protective effect of Fenofibrate in renal ischemia reperfusion injury: involved in suppressing kinase 2 (JAK2)/transcription 3 (STAT3)/p53 signaling activation[J]. Pathol Biol (Paris), 2015, 63(6):236-242.
