数字PCR和下一代测序方法用于KRAS基因突变检测中的可比性研究
2018-06-22董莲华傅博强段宇航隋志伟中国计量科学研究院北京100029
董莲华, 王 晶, 傅博强, 段宇航, 隋志伟(中国计量科学研究院, 北京 100029)
1 引 言
KRAS位于12号染色体,正常的KRAS基因参与细胞内信号传递,当KRAS基因发生突变时,会导致细胞内信号传递紊乱,从而导致细胞持续生长不能自我凋亡而引发癌变。在肿瘤患者中检测出的最常见的突变是位于KRAS基因的12、13号密码子的突变[1]。KRAS基因突变通常会发生在胰腺癌和结直肠癌形成的早期[2,3]。目前,检测KRAS突变的方法包括:基于测序技术的Sanger测序法和下一代测序法(next generation sequencing, NGS),基于聚合酶链反应(polymerase chain reaction, PCR)技术的ARMS-PCR(amplification-refractory mutation system,ARMS)、突变富集PCR[6]和COLD-PCR(co-amplification at lower denaturation temperature-PCR)。而这些方法的特异性和灵敏度有很大差异。如Sanger测序测定等位基因突变的灵敏度为20%左右[5];下一代测序方法的灵敏度为5%左右[5]; ARMS-PCR方法的灵敏度在1%[6];而突变富集PCR和COLD-PCR的灵敏度可达到0.1%[7,8]。数字PCR用于等位基因突变检测的分析灵敏度为0.05%~0.1%,甚至更高[9]。虽然对各种检测KRAS基因突变方法的报道较多,但关于不同方法之间的可比性却少有报道。
本研究的目的是建立基于数字PCR和NGS技术的KRAS基因突变检测方法,利用含有KRAS基因突变的细胞系来配置不同突变比例的样本,考察所建立的2种方法的可比性,以期为临床检测中应用这些方法时提供参考。
2 实验部分
2.1 细胞系
A549(ATCC® CRM-CCL-185TM)为携带G12S突变的纯合突变细胞,NCI-H157为携带G12R突变型的杂合突变细胞。含有野生型KRAS基因的293T细胞系作为阴性材料,用于配置含有不同突变比例的样本。
2.2 细胞培养
A549的培养基为 90%的杜尔伯科极限必需培养基(DMEM)和10%的胎牛血清(FBS);培养条件:在体积分数为5%的CO2培养箱中,温度37 ℃,培养的细胞覆盖培养皿底部面积达到80%~90%时按1:3比例传代。NCI-H157的培养基为90% 1640(培养基)+10%FBS;培养条件:在体积分数为5%的CO2培养箱中,温度37 ℃,培养的细胞覆盖培养皿底部面积达到80%~90%时按1:3比例传代。293T细胞培养液类型为90%DMEM+10%FBS培养基,在体积分数为5%的CO2培养箱中,温度37 ℃,培养的细胞覆盖培养皿底部面积达80%~90%时传代,按1:4至1:5比例传代。
2.3 不同突变比例的细胞样品的制备
2.3.1 收集细胞
NCI-H157,A549和293T细胞是贴壁细胞,使用贴壁细胞收集方法。将培养皿中的培养液吸出,每个培养皿中加入2 mL胰酶,晃动培养皿,待充分接触细胞后,将培养皿放入37 ℃培养箱中消化90 s~2 min,显微镜下观察细胞形态变圆即可;取出培养皿,加入适当培养液终止消化,将细胞悬液转移至离心管中离心,800 r/min离心8 min,弃上清,加入适当磷酸盐缓冲液,重悬细胞;然后再离心,800 r/min离心8 min离心弃上清,洗去培养液和胰酶;向离心后的细胞内加入10 mL的磷酸盐缓冲液,重新悬浮细胞得到细胞悬液。
2.3.2 细胞计数
取3支1.5 mL的离心管,分别加入900 μL的磷酸盐缓冲液,再从上述细胞悬液中分别吸取100 μL细胞液至3个离心管内,将细胞混匀,用细胞计数板进行计数。分别计算3个离心管内的细胞浓度,然后计算出每个细胞系的平均浓度。结果A549细胞浓度为8.49×106个/mL,293T细胞浓度为1.92×107个/mL,NCI-H157细胞浓度为1.69×107个/mL。
2.3.3 细胞配比混合
混合好的2种细胞的总数约为4×107个细胞,根据细胞计数计算出的细胞浓度,加入相应体积的细胞液进行混合。A549和293T混合成含有G12S不同突变比例的细胞样品见表1。NCI-H157与293T混合成含有G12R不同突变比例的细胞样品见表2。

表1 G12S不同突变比例的细胞溶液配制表

表2 G12R不同突变比例的细胞溶液配置表
2.4 数字PCR法
2.4.1 引物探针设计
本研究中选择了KRAS基因常见的突变型G12S和G12R,分别是12号染色体的25398285位置处的碱基由原来的G突变成了C(G12R)和T(G12S),从而导致由原来GGT编码的甘氨酸(Gly,G)分别变成了精氨酸(Arg,R)和丝氨酸(Ser,S),因此2个对应突变型分别标记为G12R和G12S。
根据选择的对应的突变型的序列信息,按照引物探针设计原则,使用软件设计引物探针序列。上游引物:CTGGTGGAGTATTTGATAGTGTA;下游引物:TGAAAATGGTCAGAGAAACCTTTA。野生型探针序列:VIC-tggagctGgtggcgt-MGB;G12R突变探针序列:FAM-tggagctCgtggcgt-MGB;G12S突变探针序列:FAM-ttggagctAgtggcgta-MGB。
2.4.2 PCR扩增条件及特异性检测
采用数字PCR方法测定不同突变比例的细胞样品是在芯片式数字PCR仪(BioMark,Fluidigm)上完成。对于突变比例高于1%的样品,采用48×770的芯片进行测定(每个Partition可以加入0.65 μL样品);而突变比例低于1%的样品,由于考虑到突变基因拷贝数太低,需要加大上样量,因此采用12×765的芯片(每个盘可以加入4.59 μL样品)进行测定。使用12×765的芯片,扩增的体系为10 μL,其中含上下游引物(10 μmol/L)0.5 μL,探针(10 μmol/L)0.1 μL,2.5×PCR master mix (16 S Dye,Molzym) 4 μL,DNA聚合酶(16 S Dye,Molzym)0.2 μL,GE loading (Fluidigm)0.5 μL,ROX染料 0.2 μL,用ddH2O补足。扩增条件为95 ℃,10 min,45个循环(包括95 ℃,15 s;60 ℃,40 s)。使用48×770的芯片时,所有的扩增试剂成分和浓度与上述相同,只是扩增体系降低至5 μL。
选择了7个具有常见KRAS基因突变型的DNA样本作为DNA模板,使用实时荧光定量PCR仪(LightCycler480,Roche)进行G12R和G12S PCR扩增特异性分析。荧光定量PCR扩增体系为20 μL,其中含有上下游引物(10 μmol/L)1 μL,探针(10 μmol/L)0.2 μL,2×PCR master mix 4 μL(Gene Expression,Thermo Fisher),用ddH2O补足20 μL。扩增条件为95 ℃,10 min,40个循环(包括95 ℃,15 s;60 ℃,40 s)。
2.5 下一代测序法
采用通用型柱式基因组提取试剂盒(康为世纪)将含一定突变比例的细胞样品提取基因组DNA,具体操作见试剂盒说明书。得到基因组DNA通过PCR扩增KRAS基因片段200 bp (base pair),扩增体系为50 μL,上下游引物(1 μmol/L) 各1 μL,5×HF buffer,dNTP (2.5 mmol/L)加入4.8 μL,DNA模板100 ng,Phusion超保真DNA聚合酶1 μL,水补足50 μL。使用的PCR程序为:98 ℃,2 min;35个循环:98 ℃,30 s,65 ℃,30 s,72 ℃,30 s;72 ℃,5 min。第一轮PCR程序结束后,进行第二轮PCR程序,即连接测序接头,第二轮PCR程序体系与第一轮相同,扩增程序一致,但将循环数为15个。上述2轮PCR后得到的文库采用磁珠法进行纯化,采用Qubit测定浓度,将已构建完成的文库稀释至1.5 ng/μL,进行上机测序。用实时荧光定量PCR方法(Light Cycler,Roche)对纯化后的文库进行定量,根据定量结果确定上机时所需文库量。使用NextSeq500(Illumina)进行测序。
3 结果与讨论
3.1 基因突变型的确定
为验证所使用的细胞系是否含有目标突变型以及是否为预期的纯合度,将培养后的细胞进行DNA提取和纯化后进行Sanger测序,结果见图1。图1中A5491为培养后用于配制不同不变比例的细胞样品;A5492为冻存在液氮中的细胞。用于配制不同突变比例的野生型细胞293T经过Sanger测序验证后证明在chr12-25398285位置处为野生型,且没有其它信号峰,表明该细胞系为纯合野生型。NCI-H157经过Sanger测序后确认在chr12-25398285位置同时有G和C的信号,且2个峰的峰高相近,表明此位置处为杂合突变。
A549的Sanger测序结果表明在chr12-25398285位置的主峰是T, 见图1中的A5491,即由原来野生型C突变成了T(此突变记为:T>C),但此位置同时还有一个野生型碱基C的小峰,且2个峰高相差较大。如果A549细胞是杂合,2个峰的峰高应该接近,因此推测导致这一野生型碱基峰出现的原因可能是在A549细胞培养或基因组DNA提取过程中混入了野生型细胞或DNA。为进一步证明该推测,采用NGS对其进行了验证。NGS验证结果表明A549含有83.41%的G12S突变型,同时还含有16.59%的野生型。

图1 Sanger测序法对细胞系突变型的验证结果
同时又对冻存在液氮中的A549细胞重新活化后,提取DNA进行Sanger测序验证(图1中的A5492),此次测序的结果A549为纯合突变,没有任何杂峰,因此证明第一次测序的A549样品中的确是由于混入了野生型DNA而导致了相应位置上野生型碱基峰的出现。但配置不同比例的细胞混合液时使用的是第一次测序的A549样品,因此使用该样品配制G12S不同突变比例的细胞溶液时需要考虑其纯度对配制值的影响。
3.2 PCR方法特异性
将7个具有常见KRAS基因突变型的样本DNA作为模板,采用G12R定量PCR方法进行扩增,得到的结果见图2。图2中,含有G12R突变(此突变记为:G>C)的细胞系NCI-H157样品得到了PCR扩增曲线,对应的循环数为26.37,而其它6个未含有该突变的样品没有得到相应的 PCR扩增曲线或扩增非常延迟(30个循环以内没有扩增),可见该方法对相同位点不同突变的基因没有非特异性扩增,而对目标突变的扩增特异性良好,可用于特异检测G12R突变。
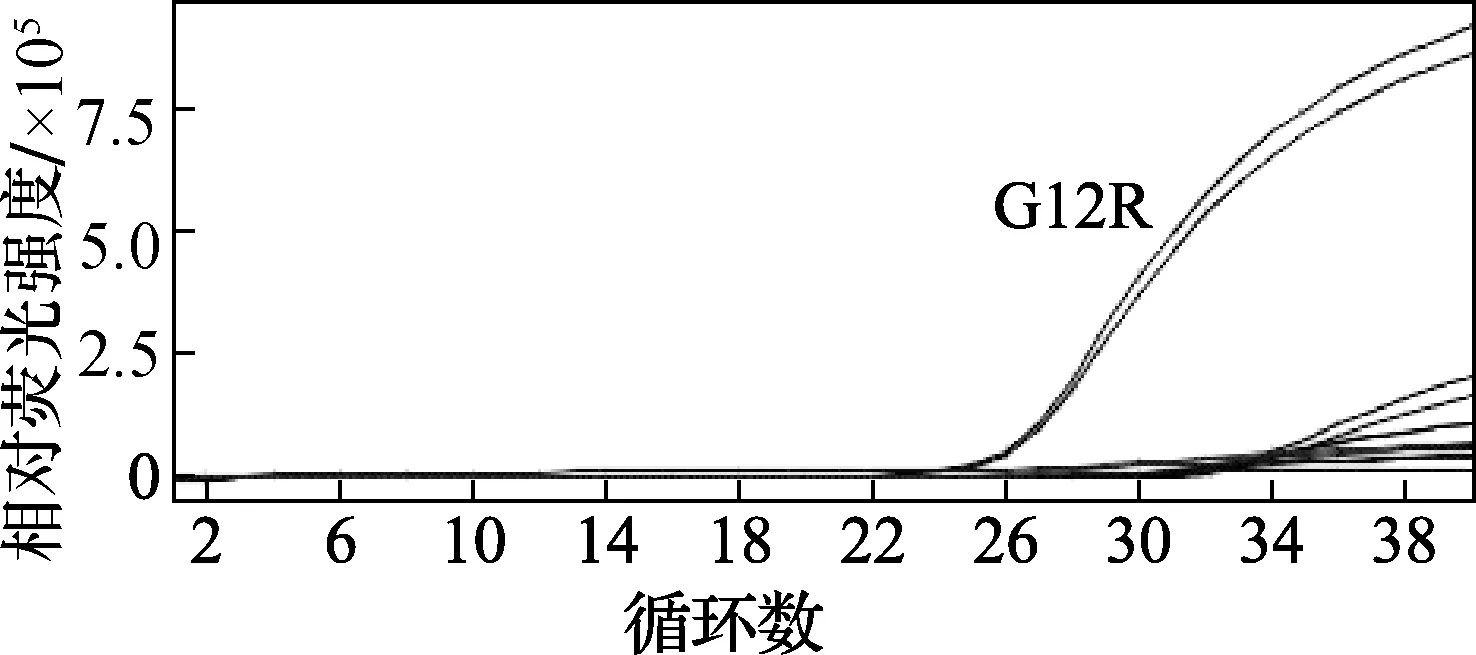
图2 G12R定量PCR方法扩增结果
将上述7个样本的DNA作为模板,进行G12S的PCR扩增特异性分析,扩增曲线见图3。由图3可知,含有G12S突变(此突变记为:G>A)的细胞系A549样品得到了PCR扩增曲线,对应的循环数为25.47,而其它6个未含有该突变的样品没有得到相应的 PCR扩增曲线,可见该方法对相同位点不同突变的基因没有非特异性扩增,而对目标突变的扩增特异性良好,可用于特异检测G12S突变。
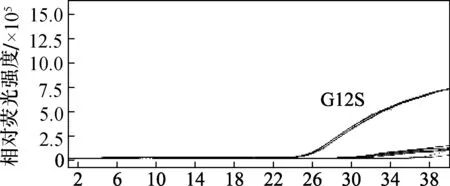
图3 G12S定量PCR方法扩增结果
3.3 数字PCR方法测定结果
基于建立的数字PCR方法对配制的2个不同突变型,每个突变型的5个不同突变比例的细胞样品进行测定。每个突变水平取5个样品,每个样品重复测定3次,G12R和G12S测定结果见表3。表3中的SD为相对标准偏差; 理论值突变丰度±SD为基于细胞计数的配制值基础上修正了突变型细胞纯度;p值为T检验的概率值。 对于未与野生型细胞混合的G12S-5和G12R-5这2个样品,数字PCR测定的结果分别为87.63%和39.59%,这与Sanger测序方法验证的结果一致,A549样品中在配制前混入了野生型细胞而导致了突变基因含量的测定结果低于100%。而NCI-H157为G12R杂合突变,但数字PCR测定的G12R突变基因含量也低于50%,基于细胞计数的配制值与数字PCR方法测定值不一致的原因可能是2种突变型细胞在配制前已经混有野生型细胞,因此基于细胞计数的突变比例配制值需要对细胞纯度进行修正,而细胞纯度参考了NGS测定结果进行修正。
数字PCR方法对突变比例高于1%的样品(G12S-3,G12S-4,G12S-5,G12R-3,G12R-4和G12R-5)的测定结果的重复性,以相对标准偏差表示为RSD<10%,测定结果与修正后的配制值相比,无显著差异。而对于突变比例低于1%的样品(G12S-1,G12S-2,G12R-1和G12R-2)的测定结果的重复性较差,RSD在16%~34%。原因是对于低突变比例样品,由于突变基因的拷贝数浓度太低,为保证野生型基因不超出12×765芯片的所能负荷的最大范围(即765个反应小室内最多有一个为阴性),加入每个小室的DNA分子数目不能超过5 000拷贝,而此时芯片中每个小室的目标突变基因拷贝数(以突变率在0.02%~0.60%计算,即每个小室的突变基因拷贝数在1~30个)在0.001~0.040拷贝/小室,这个值远远低于数字PCR所能达到的精密度最好时的值(1.59拷贝/小室)[10,11],因此在对突变基因丰度低至0.02%~0.60%的样品定量时,数字PCR方法的重复性较差。尽管如此,对于低丰度突变基因的测量,数字PCR方法仍然可以准确测定出G12S低至0.02%的突变比例,这个检测灵敏度优于Azuara等[9]报道的使用相同仪器测定KRAS基因突变时的灵敏度0.05%。
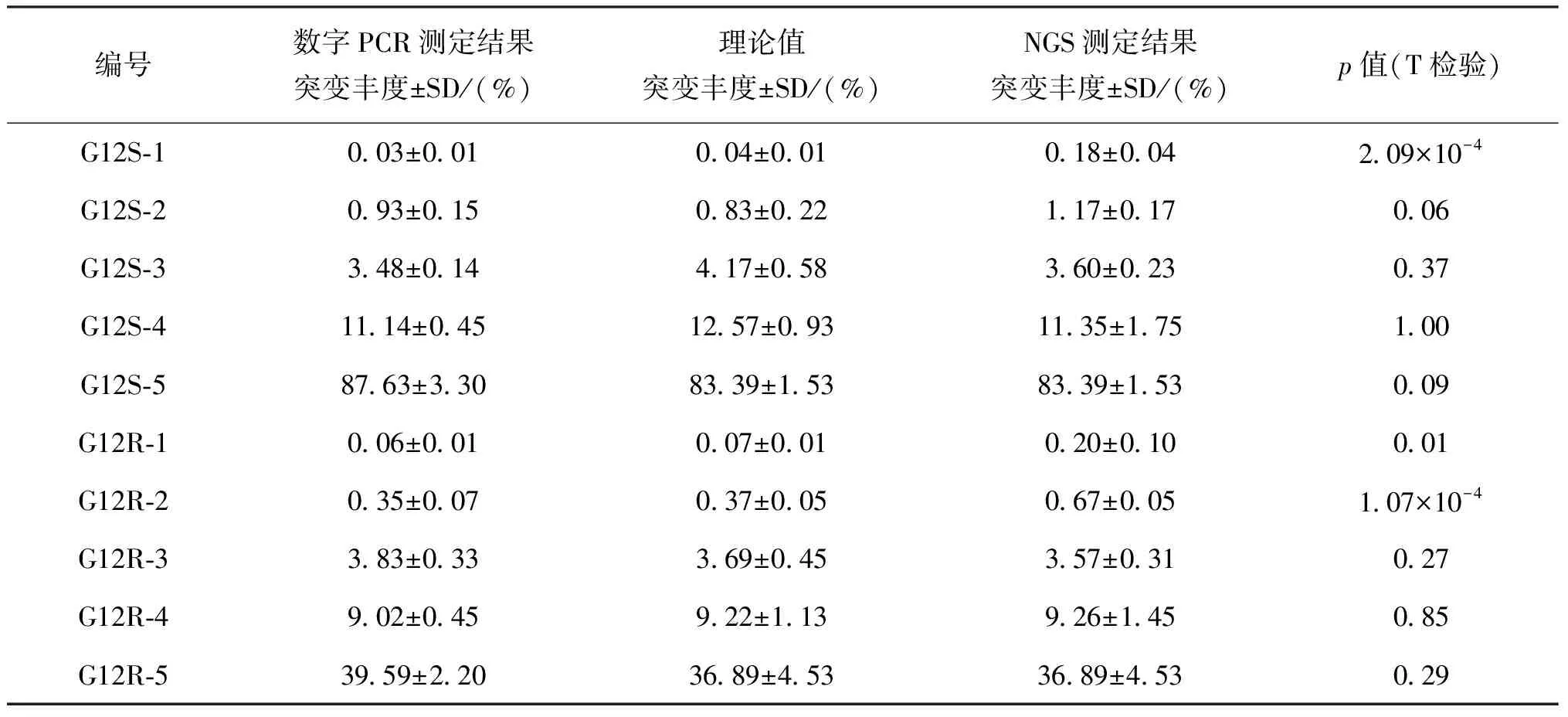
表3 突变丰度测定结果
3.4 NGS方法测定结果
采用下一代测序方法对不同突变比例的样品进行测定,每个水平的样品进行4次重复建库后测得结果见表3。对于G12S-5和G12R-5这2个未与野生型细胞混合的样品,NGS测定突变基因比例分别为83.39%和36.89%,这个结果与Sanger测序结果相符合。NGS对于突变频率高于1%的样品(G12S-3,G12S-4,G12S-5,G12R-3,G12R-4和G12R-5)的测定结果与理论值相比较无显著差异。
NGS测定的G12S-2样品的结果为1.17%,与理论值相比无明显差异。而G12R-2样品的测定结果(0.67±0.05)%与理论值相比显著偏高。NGS测定的突变比例低于0.1%的样品(G12S-1和G12R-1)的测定结果与理论值相比差异显著。而且在前2次重复中NGS方法并未检测出突变,而在后续的2次重复中随着测序深度的增加,可以检测出突变型了,但是测定结果仍与理论值差距较大,这表明该突变比例已经完全超出了NGS所能测定出的突变水平,而多数报道指出NGS所能测定突变比例一般为5 %[5],本研究中所建立的NGS方法将测序深度增加至105数量级,可以检测低至1 %左右的突变。
3.5 2种方法的可比性
对NGS和数字PCR这2种方法的测定结果,采用T检验进行统计学差异显著性判断,分析结果见表3。对于突变比例≥0.83%的样品,NGS和数字PCR这2种方法的测定结果均没有显著差异(p>0.05)。而对于突变比例低于0.83 %的样品(G12S-1,G12R-1,G12R-2),2种方法的测定结果差异显著(p<0.05)。
4 结 论
从NGS几次重复测定结果可以看出本研究中所建立的针对KRAS基因突变的NGS方法的最低检测突变比例约为1%,当突变比例等于或高于NGS方法的测定灵敏度时,数字PCR方法和NGS方法的测量结果可比,而当突变比例低于NGS方法的测定灵敏度时,数字PCR方法和NGS方法的测量结果不可比。
[参考文献]
[1] Russo A, Bazan V, Agnese V,etal. Prognostic and predictive factors in colorectal cancer: Kirsten Ras in CRC (RASCAL) and TP53CRC collaborative studies[J].AnnalsofOncology,2005, 16(Supplement 4): iv44-iv49.
[2] Esteller M, Gonzaález S, Risques R A,etal. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer[J].JournalofClinicalOncologyOfficialJournaloftheAmericanSocietyofClinicalOncology, 2001,19(2):299 -304.
[3] Kisiel J B,Yab T C, Taylor W R,etal. Stool DNA testing for the detection of pancreatic cancer: assessment of methylation marker candidates[J].Cancer, 2012,118(10):2623-2631.
[4] Bagci P, Andea A A, Basturk O,etal. Large duct type invasive adenocarcinoma of the pancreas with microcystic and papillary patterns: a potential microscopic mimic of non-invasive ductalneoplasia[J].ModernPathology, 2011, 25(3):439- 448.
[5] Ibrahem S, Seth R, O’Sullivan B,etal. Comparative analysis of pyrosequencing and QMC-PCR in conjunction with high resolution melting for KRAS/BRAF mutation detection[J].InternationalJournalofExperimentalPathology, 2010,91:500- 505.
[6] Linardou H, Briasoulis E, Dahabreh I J,etal. All about KRAS for clinical oncology practice: gene profile, clinical implications and laboratory recommendations for somatic mutational testing in colorectal cancer[J].CancerTreatmentReviews, 2011,37(3):221-233.
[7] Li J,Makrigiorgos G M. A new platform for highly improved mutation detection in cancer and genetic testing[J].BiochemicalSocietyTransactions,2009,37(Pt 2): 427-432.
[8] Milbury C A, Li J, Makrigiorgos G M. Ice-COLD-PCR enables rapid amplification and robust enrichment for low-abundance unknown DNA mutations[J].NucleicAcidsResearch,2011,39(1):1-10.
[9] Azuara D, Ginesta M M, Gausachs M,etal.Nanofluidic digital PCR for KRAS mutation detection and quantification in gastrointestinal cancer[J].ClinicalChemistry,2012: 58(9):1332-1341.
[10] Huggett J F, Foy C A, Benes V,etal. The Digital MIQE Guidelines:Minimum Information for Publication of Quantitative Digital PCR Experiments[J].ClinicalChemistry, 2013,59(6):892-902.
[11] 董莲华,隋志伟,王晶,等. 数字PCR方法准确测量质粒DNA拷贝浓度[J].计量学报,2017, 38(2):247-251.
