CDC25B与CDK2/Cyclin A蛋白相互作用研究
2018-05-30陈秀博晋文燕吴晓辉马英
陈秀博,晋文燕,吴晓辉,马英
(天津医科大学药学院,天津市临床药物关键技术重点实验室,天津300070)
CDC25B是细胞周期的重要调控蛋白,它是调节CDK-cyclin复合物活性的重要因子[1]。由于其调节作用,CDC25B与癌症紧密相连,也成为治疗癌症的潜在靶点。CDK-cyclin复合物在细胞周期调控中起核心作用,当Cyclin A和CDK相结合后,CDK分子暴露了14位和15位的苏氨酸和酪氨酸,激酶Weel能对14位和15位的苏氨酸和酪氨酸进行磷酸化,从而抑制了CDK的活性[2]。CDC25B-CDK2/Cyclin A复合物的晶体结构还未见报道。本研究旨在研究CDC25B-CDK2/Cyclin A复合物的结合模式,经过分析其关键结合位点,阐明了CDC25B与CDK2/Cyclin A蛋白间参与相互作用的残基,为新型CDC25B抑制剂的设计提供理论依据。
1 材料与方法
1.1 蛋白-蛋白对接 蛋白-蛋白对接是基于两个已知蛋白的三维结构,通过分子模拟方法预测复合物的近天然结构。使用Discovery Studio v3.5中的ZDOCK[3-5]和RDOCK[6]模块来实现蛋白与蛋白的对接计算。ZDOCK是一种基于快速傅里叶转化相关性技术的刚性蛋白对接算法。RDOCK是一种基于CHARMm的能量优化过程,用于优化ZDOCK所寻找到的蛋白-蛋白复合物的结合构型,并使用能量打分函数给这些结合构型打分。
选取CDC25B蛋白作为对接的受体(PDB ID:4WH9)[7],将从晶体库[8]中下载下来的 CDK2/Cyclin A 的晶体结构(PDB ID:5CYI)[9]作为对接的配体。首先使用 Discovery Studio v3.5 中“prepare protein”[10-11]对受体蛋白和配体蛋白进行结构优化,包括去掉结晶水和处理二硫键,处理金属离子,按照预期的温度和pH为蛋白分子加上末端氢原子。然后使用Discovery Studio v3.5中的ZDOCK模块预测CDC25B与CDK2/Cyclin A的复合物结构。设置旋转采样的配体方向欧拉角度步长为6,“RMSD Cutoff”为 6.0,“Interface Cutoff”为 9.0,“Maximum Number of Clusters”为 60,结合模式“Top Poses”为2000,其它参数均使用缺省值,进行计算。采用ZRANK方法对ZDOCK对接得分进行重新排序。最后,基于CHARMM polar H力场采用RDOCK方法对ZDOCK结果进行重新优化和打分,选出前五个E_RDOCK得分较低的构象进行下一步的分析。使用superimpose程序将通过RDOCK优化后的5个结合模式中的CDC25B蛋白和CDK2/Cyclin A蛋白与晶体库中已有蛋白的构象进行叠合比较,选出RMSD <3Å[12]的构象。
1.2 预测CDC25B蛋白与CDK2/Cyclin A蛋白的结合位点 蛋白-蛋白复合物是通过蛋白质间的非共价相互作用形成的,其主要组成部分包括疏水相互作用、氢键相互作用、静电相互作用等。疏水相互作用是指由于非极性基团的存在,分子中极性基团的静电力和氢键力基团有聚集在一起的倾向而对疏水基团产生排斥,从而导致水分子结构重排,水和水之间形成氢键网络,造成疏水基团相互聚拢所产生的能量效应和熵效应。氢键相互作用是指由一个缺电的H原子和一个电负性强的原子或原子团X(如F、O、N等)之间形成的一种弱相互作用。应用“Analyze Protein Interface”模块计算CDC25B蛋白和CDK2/Cyclin A蛋白的溶剂可及表面积(SAS)并分析CDC25B蛋白和CDK2/Cyclin A蛋白相互作用界面的关键氨基酸残基。“Calculate Interaction Energy”模块计算CDC25B蛋白和CDK2/Cyclin A蛋白相互作用界面处的关键氨基酸残基间的相互作用能。通过分析CDC25B蛋白与CDK2/Cyclin A蛋白结合界面处关键氨基酸残基的相互作用预测CDC25B蛋白与CDK2/Cyclin A蛋白的结合位点。
2 结果
2.1 蛋白-蛋白对接的研究
2.1.1 获得CDC25B-CDK2/Cyclin A复合物 本实验应用ZDOCK和RDOCK方法模拟得到CDC25B与CDK2/Cyclin A的复合物结构。ZDOCK算法是基于傅立叶快速转换技术的大分子刚性对接算法,并且充分考虑了配体蛋白的柔性,在蛋白-蛋白的对接中具有很高的准确性;而RDOCK算法是基于CHARMM力场能量的最小化并应用静电势能和去溶剂化能组成的自由能打分函数来预测结合位点准确性的方法。
通过ZDOCK算法共产生了60簇包括2 000个构象并且通过ZDOCKScore进行排名,选取前100个结构进行后续优化。应用RDOCK算法对选取的复合物结构进行优化,根据E_DOCK Score进行排名。E_RDOCK Score得分越低,表明该pose对接结果越好,越接近真实的对接构象。选出前5个E_RDOCK Score较低的构象(表1),应用superimpose程序将这5个构象中的CDC25B和CDK2/CyclinA蛋白与晶体库中已有蛋白的构象进行叠合。将这5个构象中的CDC25B蛋白与此蛋白的构象进行叠合后,其RMSD 值分别为 0.32Å,0.40Å,0.35Å,0.44Å,0.36Å。CDK2/Cyclin A蛋白的RMSD值分别为0.40Å,0.38 Å,0.42Å,0.34Å,0.43Å。RMSD 值越小,代表其构象越接近真实结构的构象。因此,选择pose 1(图1A)为目标对象分析CDC25B蛋白与CDK2/Cyclin A蛋白结合界面处关键氨基酸残基的相互作用,预测CDC25B蛋白与CDK2/Cyclin A蛋白的结合位点。

表1 ZDOCK和RDOCK对接后得分Tab 1 Docking score(ZDOCK and RDOCK)

图1 (A)CDC25B蛋白与CDK2/Cyclin A蛋白对接模式图;(B)CDC25B蛋白与CDK2/Cyclin A蛋白结合界面处关键氨基酸残基相互作用图Fig 1 (A)The docking model of CDC25B and CDK2/Cyclin A;(B)The interaction of key residues in CDC25B and CDK2/Cyclin A surface
2.2 CDC25B-CDK2/Cyclin A复合物相互作用分析 以上一步得到的最优结构pose 1为目标,应用“Analyze Protein Interface”和“Calculate Interaction Energy”模块分析CDC25B-CDK2/Cyclin A复合物之间的相互作用。
2.2.1 疏水相互作用分析 疏水/亲水间的平衡是蛋白质结构的重要特征,蛋白质分子内部的疏水作用在一定程度上决定了其结构稳定性,它是支撑蛋白质空间结构稳定必不可少的部分,对蛋白质的结构和功能有极其重要的影响。图2是CDC25BCDK2/Cyclin A复合物疏水性氨基酸残基的分布图,蓝色为亲水性氨基酸残基,棕色为疏水性氨基酸残基,颜色深浅和亲疏水性大小呈正相关,亲水性到疏水性的区间为-3.0至3.0。表2和表3列出了CDC25B-CDK2/Cyclin A复合物界面处所含极性残基和非极性残基的SAS值。统计分析表明CDC25B蛋白和CDK2/Cyclin A蛋白结合界面处的Polar Contact Surface Area 值分别为 396.89 Å2和429.32 Å2,Nonpolar Contact Surface Area 值分别为429.32 Å2和 376.70 Å2,应用半经验法对 CDC25BCDK2/Cyclin A复合物的疏水率(hydrophobicity)进行定量描述,

其中NONSAS和POLSAS分别表示 HNRPKAGPS复合物结合界面处非极性基团和极性基团的溶剂可接近表面积总和,下标1和2分别表示CDC25B蛋白和CDK2/Cyclin A蛋白,代入上述公式计算得到CDC25B蛋白和CDK2/Cyclin A蛋白之间的疏水率为48.88%。CDC25B蛋白中GLY380,PHE475,ARG490,LEU540,ARG544,LEU545 氨基酸残基和 CDK2/Cyclin A 蛋白中 LYS9,LEU37,ILE49,ILE52,LEU54,VAL69,THR72,GLU73,LEU76,LYS89,ASP92,LYS129,ARG150,ALA151,PHE152,THR165,ARG169,SER181,PHE213,ARG214,ARG217(CDK2),VAL175,ASP177,LEU186,MET189,GLU230,LEU263,GLU274,LYS288,LEU292,ARG293,GLU295,LEU299,ASP305,LEU320(Cyclin A)氨基酸残基表现出了疏水性质,使两蛋白间形成了较强的疏水界面。
2.2.2 氢键和共轭作用分析 氢键和共轭作用是分子相互作用中重要的作用力,对整个系统的稳定起着重要的作用。图1B所示,CDC25B-CDK2/Cyclin A复合物相互作用界面处共含有1个Pi相互作用和6个氢键来维持其三维结构的稳定。CDC25B:ARG485 的 NH 和 CDK2/Cyclin A:TRP167形成Pi-Cation相互作用。表4列出了CDC25B蛋白和CDK2/Cyclin A蛋白结合界面处所有参与氢键形成的氨基酸残基及其氢键键长,它们主要是由一侧亚基上氨基酸残基的羰基氧和另一侧亚基氨基酸残基的酰胺氢或巯基氢相互作用所形成。由表4可知结合界面处有4对氨基酸残基间(TYR382:OH-TYR382:HH;ASP206:OD2-ASP210:OD1;ARG488:HH-ASP206:O;ARG492:HH-ASP206:OD2;ARG492:HE-ARG492:HH;SER207:OG-ASP210:OD2)的氢键键长均小于2.7Å,属于短强氢键,是稳定CDC25B-CDK2/Cyclin A复合物结构的主要作用力。
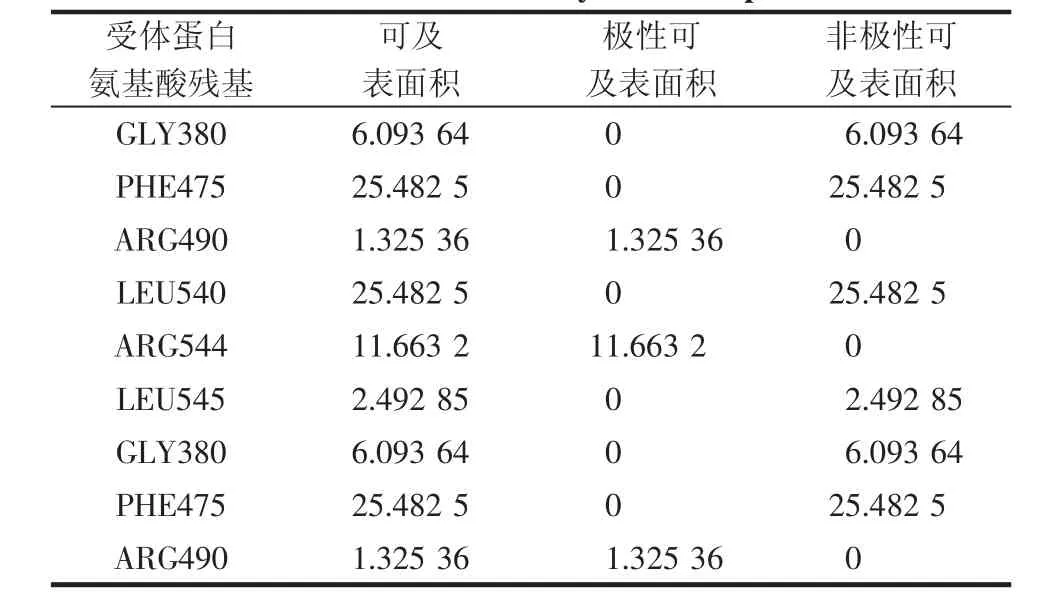
表2 CDC25B-CDK2/Cyclin A复合物界面处CDC25B极性残基和非极性残基的SAS值Tab 2 The SAS of CDC25B polar and nonpolar residues in the surface of CDC25B-CDK2/Cyclin A complex

表3 CDC25B-CDK2/Cyclin A复合物界面处CDK2/Cyclin A极性残基和非极性残基的SAS值Tab 3 The SAS of CDK2/Cyclin polar and nonpolar residues in the surface of CDC25B-CDK2/Cyclin A complex

图2 CDC25B-CDK2/Cyclin A复合物结合界面处的疏水分布Fig 2 Distribution of hydrophobic of CDC25B-CDK2/Cyclin A complex surface

表4 CDC25B-CDK2/Cyclin A复合物结合界面处残基间氢键统计Tab 4 Hydrogen bond statistics of CDC25B-CDK2/Cyclin A complex surface
2.2.3 相互作用能分析 为了确定CDC25B蛋白和CDK2/Cyclin A蛋白相互作用中起作用的重要氨基酸残基,本实验在CHARMm力场下计算了CDC25B蛋白和CDK2/Cyclin A蛋白位于界面处的各氨基酸残基的相互作用能。计算结果显示CDC25B蛋白和CDK2/Cyclin A蛋白的总相互作用能为-41.697kcal/mol,其中范德华(VDW)作用能和静电作用能分别为-14.219kcal/mol和-27.478kcal/mol,静电作用能明显大于VDW作用能,这说明静电作用是促使复合物结构形成的主要驱动力。表5列出了CDC25B-CDK2/Cyclin A结合界面处重要氨基酸残基的相互作用能。在CDC25B蛋白的结合界面处的 氨 基 酸 残 基 包 括 GLY380,TYR382,ARG485,ARG488,GLU489,ARG490,ARG492,TYR497,在CDK2/CyclinA蛋白的结合界面处的氨基酸残基包括THR165,TRP167,ASP206,SER207,ASP210,PHE213,它们之间的相互作用能对CDC25B-CDK2/CyclinA复合物活性区域结构的稳定起着重要作用。以上这些分析揭示了CDC25B蛋白和CDK2/Cyclin A蛋白的结合模式及起相互作用的氨基酸残基,为之后研究CDC25B蛋白与CDK2/Cyclin A相互作用奠定基础。

表5 CDC25B-CDK2/Cyclin A复合物结合界面处残基的相互作用能统计Tab 5 Interaction statistics of CDC25B-CDK2/Cyclin A complex surface
3 讨论
本实验通过蛋白-蛋白对接方法模拟了CDC25B-CDK2/Cyclin A复合物的空间结构,并应用“Analyze Protein Interface”和“Calculate Interaction Energy”模块分析两蛋白之间的相互作用。应用ZDOCK和RDOCK的方法,对CDC25B蛋白与CDK2/Cyclin A蛋白进行对接,并根据对接得分及superimpose程序,挑选出得分最高,RMSD值最小的构象(pose 1)。最后应用“Analyze Protein Interface”和“Calculate Interaction Energy”模块,对pose 1的疏水相互作用,氢键相互作用,相互作用能进行计算分析,预测得到CDC25B-CDK2/Cyclin A复合物的结合模式及起相互作用的氨基酸残基(CDC25B:GLY380,TYR382,ARG485,ARG488,GLU489,ARG490,ARG492,TYR497;CDK2/CyclinA:THR165,TRP167,ASP206,SER207,ASP210,PHE213)。为之后研究CDC25B蛋白与CDK2/Cyclin A蛋白相互作用奠定了基础,同时也为抗癌药物的研究发展提供了新的思路。
[1]Kristja’nsdo’ttir K,Rudolph J.Cdc25 phosphatases and cancer[J].Chem Biol,2004,11(8):1043
[2]MorganOD.Principal of CDKregulation[J].Nature,1995,374(6518):131
[3]Wiehe K,Pierce B,Mintseris J,et al.ZDOCK and RDOCK performance in Capri rounds 3,4,and 5[J].Proteins,2005,60(2):207
[4]Chen R,Li L,Weng Z P.ZDOCK:an initial-stage protein-docking algorithm[J].Proteins,2003,52(1):80
[5]Chen R,Weng Z P.A novel shape complementarity scoring function for protein-protein docking[J].Proteins,2003,51(3):397
[6]Li L,Chen R,Rdock W Z.Refinement of rigid-body protein docking predictions[J].Proteins,2003,53(3):693
[7]Lund G,Dudkin S,Borkin D,et al.Inhibition of CDC25B phosphatase through disruption of protein-protein interaction[J].ACS Chem Biol,2015,10(2):390
[8]Zardecki C,Dutta S,Goodsell D S,et al.RCSB protein data bank:a resource for chemical,biochemical,and structural explorations of large and small biomolecules[J].J Chem Educ,2016,93(3):569
[9]Martin M,Anscombe E,Meschini E,et al.Identification and characterization of an irreversible inhibitor of CDK2[J].Eur J Cancer,2014,50(6):106
[10]Spassov V Z,Flook P K,Yan L S.LOOPER:a molecular mechanics-based algorithm for protein loop prediction[J].Protein Eng Des Sel,2008,21(2):91
[11]Spassov V Z,Yan L.A fast and accurate computational approach to protein ionization[J].Protein Sci,2008,17(11):1955
[12]Kingsley L J,Esquivel-Rodriguez J,Yang Y A,et al.Ranking protein-protein docking results using steered molecular dynamics and potential of mean force calculations[J].J Comput Chem,2016,37(20):1861
