HSP22在高脂致动脉粥样硬化病变中的作用及对他汀干预的影响*
2018-05-17吴延庆
朱 敏, 吴延庆
(南昌大学第二附属医院心血管内科, 江西 南昌 330006)
动脉粥样硬化(atherosclerosis,AS)是一个慢性炎症反应过程。在这个过程中内皮损伤起着关键作用。高脂是内皮损伤的始动因素,尤其是氧化型低密度脂蛋白(oxidized low-density lipoprotein,ox-LDL)[1]。有研究表明,ox-LDL通过增加活性氧簇(reactive oxygen species,ROS)的产生,加重细胞氧化损伤;同样ox-LDL可促进黏附分子及热休克蛋白(heat shock proteins,HSPs)的表达,抑制内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)和前列腺素的产生,促进内皮细胞分泌黏附分子,如细胞间黏附分子1(intercellular adhesion molecule-1,ICAM-1)和血管细胞黏附分子1(vascular cell adhesion molecule-1,VCAM-1)等,从而诱导炎症反应,故抗炎是治疗AS的重要途径之一[2-3]。
HSPs是一类生物体在各种应激状态下产生的结构上高度保守的蛋白,其可使受损蛋白恢复正常结构和功能并且抵御有害刺激,并促进细胞存活和适应外界环境,HSPs根据分子量大小可分为HSP110、HSP90、HSP70、HSP60、HSP40、小分子热休克蛋白家族(small heat shock proteins, sHSPs)及泛素[4]。有研究表明HSPs能够影响AS的发生发展,如HSP27通过抑制组织氧化应激,降低炎症免疫反应,从而抑制血管平滑肌的增殖、迁移及抗凋亡等效应起到抗AS的作用[5];而HSP60通过诱导抗HSP60适应性免疫反应加重内皮功能紊乱,导致AS炎症反应[6];HSP90则通过降低一氧化氮(nitric oxide,NO)的产生,从而减少eNOS和增加氧自由基的产生,加重了内皮功能的紊乱[7];HSP70在血管受应激时保护血管免于受损,并通过促进抗炎因子白细胞介素(interleukin,IL)-10的表达来调节免疫反应[8]。但在某些情况下,HSP70又是有害因子,例如有研究表明高脂环境可以诱导HSP70表达升高,用外源性的HSP70孵育单核细胞可增加单核细胞与内皮细胞的黏附,并大量释放IL-6,促进内皮细胞表达ICAM-1,进一步加重AS的进展[9]。
sHSPs是分子量12~43 kD的HSPs,均具有ATP激酶活性,其C端都有一段长约80~100个氨基酸的高度保守结构域,称为a晶体结构域。热休克蛋白22(heat shock protein 22,HSP22)是sHSPs家族成员之一,分子量为21.6 kD,又称为HSPB8、E21G1、a晶状体C和H11[10]。与其它sHSPs家族成员一样具有分子伴侣和自身激酶等生物活性,可调节细胞增殖、迁移并抑制细胞凋亡,对细胞起到保护性作用。Marunouchi等[11]通过对心梗后心衰模型的研究发现,在心梗后心衰代偿期心肌细胞HSP22的表达增加,并保护线粒体功能,而在失代偿期,HSP22的减少与线粒体功能减退呈正相关,并加速了心衰的进展,表明HSP22对心肌细胞有保护性作用。我们团队前期研究发现在ox-LDL诱导的内皮损伤模型中,HSP22可通过抑制p38丝裂原激活蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路的激酶,激活细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)信号通路,减少细胞凋亡,同时降低肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)的表达,减轻炎症反应,促进eNOS表达,说明HSP22具有抗氧化应激及抗炎作用,延缓了AS的进展[12]。同时我们也构建了大鼠AS模型,发现HSP22及TNF-α在高脂血症大鼠主动脉的表达升高,而eNOS的表达下降,他汀干预可降低HSP22及TNF-α的表达[13]。
根据前期研究结果,我们推测HSP22在高脂致内皮损伤中发挥保护作用,而他汀的保护作用可能是通过对HSP22的调节而产生。在本研究中,我们通过构建HSP22基因缺失及过表达的ApoE-/-小鼠,并给予高脂饮食建立AS模型,探讨HSP22基因在高脂致AS病变中的作用及对阿托伐他汀(atorvastatin,Ator)干预的影响。
材 料 和 方 法
1 动物和主要试剂
8~9周龄、体重20~24 g的SPF级ApoE-/-、HSP22-/-ApoE-/-和HSP22+ApoE-/-雄性小鼠各18只,其中ApoE-/-雄性小鼠购于苏州工业园区爱尔麦特科技有限公司(合格证编号为2014-0007),HSP22-/-ApoE-/-及HSP22+ApoE-/-雄性小鼠购于北京维通达生物技术有限公司(合格证编号为11806300000247)。动物饲养于南昌大学转化医学研究院SPF级动物饲养房,配有S8智能型独立通气笼IVC系统,笼具及饮用水均灭菌消毒处理,光照及黑暗时间各12 h,温度21 ℃,湿度77.5%。实验动物的饲养和处理程序经南昌大学第二附属医院医学研究伦理委员会审查通过,审批号为研临审【2012】第(002)号。普通饲料为小鼠繁殖饲料(SPF级),高脂饲料为乳脂动脉粥样硬化饲料(主要成分为蛋白质20%、碳水化合物 50%、脱水乳脂21%和胆固醇 0.15%),购于江苏美迪森生物医药有限公司;阿托伐他汀钙片购于辉瑞制药有限公司。
2 实验方法
2.1实验分组及动脉粥样硬化模型的建立 3种小鼠适应性基础饲料喂养1周后,称重,每种小鼠按体重由小到大排序,随后采用随机数字表分别将3种小鼠随机分为对照(control)组与Ator干预组(Ator)组,HSP22基因缺失(HSP22 knockout,KO)组与HSP22基因缺失Ator干预(KO+Ator)组及HSP22过表达(HSP22 overexpression,Tg)组与HSP22过表达Atro干预(Tg+Ator)组,均高脂饮食12周,其中Ator组、KO+Ator组及Tg+Ator组从第5周开始给予Ator (10 mg·kg-1·d-1)干预,所有对照组给予等量生理盐水干预,共饲养13周,建立动脉粥样硬化模型。
2.2取材 在禁食不禁饮12 h 后,10%水合氯醛0.05~0.07 mL腹腔麻醉,眼内眦静脉采血法采集适应性喂养1周后及干预结束后小鼠的空腹血,离心取血清。干预结束取血后,迅速剪开胸腹腔,用预冷的PBS冲洗并分离出心脏及整个主动脉。每组取6只小鼠的主动脉根部(包括主动脉瓣环、主动脉瓣尖和主动脉窦)血管用于HE染色,每组取6只小鼠的主动脉弓(头臂干起始部至主动脉峡部)用于免疫组化,每组取3只小鼠整个主动脉(头臂干起始部至髂总动脉分叉处整条动脉)用于整体油红O染色。其余血管组织用于蛋白定量分析。
2.3血清学指标的检测 根据试剂盒说明书测定干预前及干预结束后血清中甘油三酯(triglyceride, TG)、总胆固醇(total cholesterol,TC)、高密度脂蛋白胆固醇(high-density lipoprotein cholesterol,HDL-C)和低密度脂蛋白胆固醇(low-density lipoprotein cholesterol, LDL-C),其中TC和TG检测采用酶法分析,LDL-C和HDL-C采用选择性沉淀法测定(BioSino)。同时采用酶联免疫吸附实验检测血清中HSP22和IL-6的水平(RayBiotech)。
2.4主动脉斑块的形态学观察 将主动脉根部进行常规石蜡包埋,切片,取经主动脉瓣膜处动脉组织进行HE染色,经脱蜡、水化、染色、脱水、二甲苯透明、中性树胶封片,显微镜拍照。清除整个主动脉外膜脂肪组织,4%多聚甲醛固定过夜,PBS冲洗1 d 后,异丙醇脱水数分钟,室温下经油红O染色4 h(避光),将动脉取出后在85%异丙醇中冲洗4次,每次5min,PBS冲洗干净并将动脉伸展开,行数码拍照。
2.5Western blot法检测HSP22、eNOS、核因子κB(nuclear factor-κB,NF-κB)及ICAM-1的蛋白表达 取出动脉组织置于冰上,称重,加入液氮研磨,在研磨好的组织中加入裂解液(1 mL RIPA裂解液+10 μL PMSF+10 μL的磷酸酶抑制剂),裂解60 min后离心取上清得到样品,并用BCA法测定蛋白浓度。蛋白在SDS-PAGE中分离,并转移到PVDF膜上,用含5%脱脂奶粉的TBST封闭2 h,结合Ⅰ抗(HSP22,1∶500,Abcam;eNOS,1∶500,Abcam;NF-κB,1∶1 000,Affinity Biosciences Company;ICAM-1,1∶1 000, Cell Signaling Technology; GAPDH,1∶1 000,中杉金桥生物技术公司), 4 ℃过夜; TBST漂洗3次; Ⅱ抗(兔抗、鼠抗均为1∶5 000)室温孵育2 h,TBST漂洗3次;加ECL发光液显色,在Bio-Rad凝胶成像系统中观察并拍照。
2.6免疫组化法检测主动脉中HSP22和NF-κB的表达水平 主动脉弓血管组织切片脱蜡后,置于PBS中,经抗原修复液高温修复,Tris-HCl缓冲盐溶液清洗2次,3%过氧化氢溶液室温10 min,清除内源性过氧化物酶,Tris-HCl缓冲盐溶液清洗3次,血清封闭液封闭1 h,滴加HSP22单克隆抗体(1∶100)和NF-κB多克隆抗体(1∶50)4 ℃过夜,添加Ⅱ抗1 h,DAB 染色,苏木精复染,中性树胶封片,显微镜拍照。
3 统计学处理
采用SPSS 13.0统计软件对数据进行统计分析,计量资料以均数±标准差(mean±SD)表示,多组间比较用单因素方差分析,方差分析后各组均数间的两两比较应用SNK-q检验。以P<0.05为差异有统计学意义。
结 果
1 小鼠基因型鉴定结果
图1A为ApoE基因缺失小鼠鉴定结果,仅在245 bp处出现透亮条带者为ApoE-/-小鼠;图1B为HSP22基因缺失小鼠鉴定结果,仅在175 bp处出现透亮条带者为HSP22-/-小鼠,在371 bp及175 bp均出现条带者为HSP22+/-,仅在371 bp处出现条带者为HSP22野生型小鼠;图1C为HSP22过表达小鼠鉴定结果,在241 bp处出现透亮条带者为HSP22+小鼠。后续实验挑选基因型符合的小鼠。

Figure 1. The genotyping results ofApoE-/-mice (A),HSP22-/-mice (B) andHSP22+mice (C) identified by PCR.
图13种小鼠的PCR基因型鉴定结果
2 小鼠体重的变化
适应性喂养1周后,小鼠按体重分层随机分组,各组间基线体重的差异无统计学显著性。在高脂饮食4周后(干预前),各组间小鼠体重的差异无统计学显著性。各组小鼠继续喂食高脂饲料8周,其中Ator组、KO+Ator组及Tg+Ator组从第5周开始给予Ator干预。干预结束后,Ator组小鼠体重低于对照组(P<0.05),Tg+Ator组低于Tg组(P<0.05);其中Tg+Ator组及Ator组小鼠体重均低于KO+Ator组(P<0.05),对照组及KO组均高于Tg组(P<0.05),Ator组与Tg+Ator组及对照组与KO组体重的差异无统计学显著性,见图2。
3 血脂水平的变化
3.1基线血脂 适应性喂食1周后,各组间基线血脂水平的差异均无统计学显著性,见表1。
3.2干预结束时血脂的变化 经高脂饲料饮食12周后,Tg组血清LDL-C及TC较KO组降低(P<0.05),对照组、KO组与Tg组小鼠血清中TG和HDL-C的差异无统计学显著性。给予Ator干预8周后,Ator组血清TC及LDL-C低于对照组(P<0.05),HDL-C高于对照组(P<0.05);KO+Ator组的血清TC及LDL-C较KO组明显降低(P<0.05),HDL-C高于KO组(P<0.05);Tg+Ator组血清TC及LDL-C较Tg组明显降低(P<0.05),HDL-C高于Tg组(P<0.05);其中Tg+Ator组血清中LDL-C和HDL水平与Ator组及KO+Ator组的差异无统计学显著性。各组小鼠血清中TG变化的差异无统计学显著性,见图3。
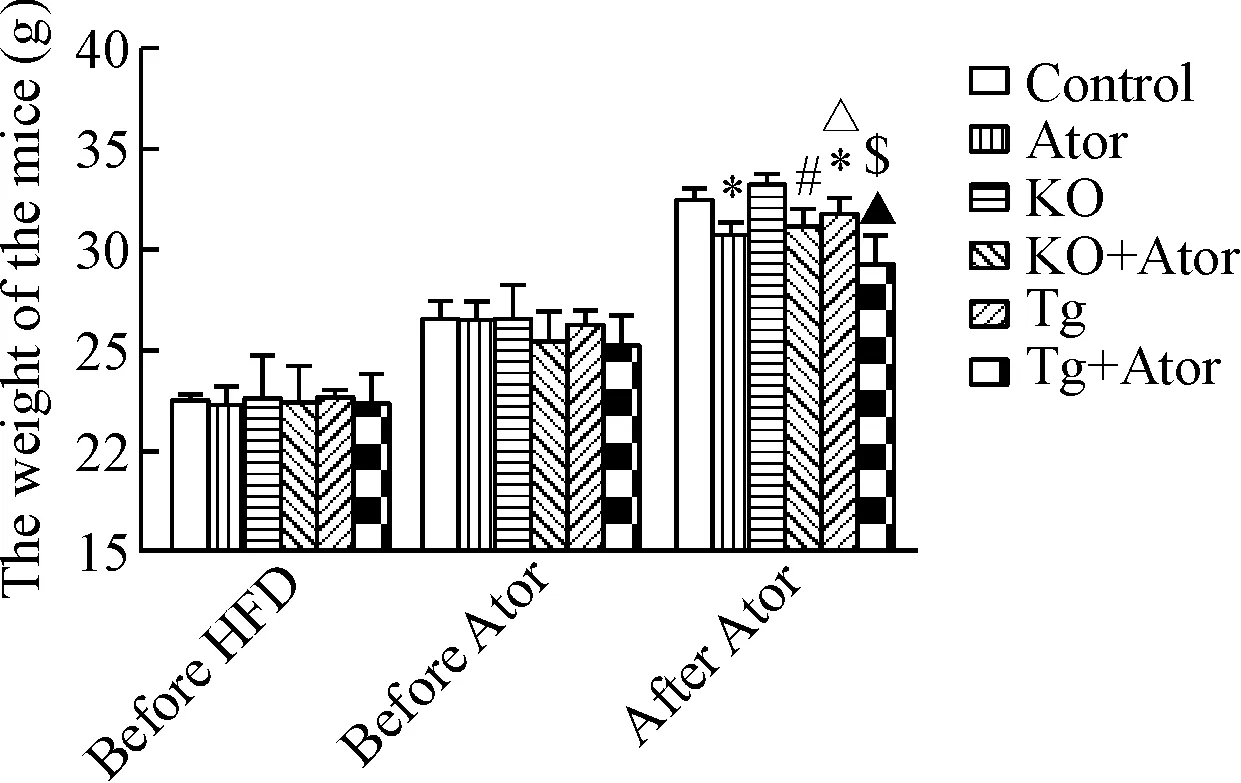
Figure 2. The body weight of the mice before and after treatment with high-fat diet (HFD) and atorvastatin (Ator). Mean±SD.n=9.*P<0.05vscontrol group;#P<0.05vsAtor group;△P<0.05vsKO group;▲P<0.05vsKO+Ator group;$P<0.05vsTg group.
图2各组小鼠基线、干预前及干预后体重的变化
4 病理形态学的变化
4.1主动脉整体油红O染色 干预结束后,行主动脉整体油红O染色,结果显示对照组、KO组及Tg组红染脂质斑块相对较多,在主动脉弓、腹主动脉及发出颈动脉处有多处斑块;而Ator干预8周后斑块面积较单纯高脂饮食减少,见图4A。比较小鼠主动脉整体油红O染色的斑块相对面积(斑块总面积/血管总面积),结果显示,Ator组较对照组减少(P<0.05),KO+Ator组较KO组减少(P<0.05),Tg+Ator组较Tg组减少(P<0.05),见图4B。
表1血脂基线水平的检测结果
Table 1. Blood lipid levels of the mice before treatment with high-fat diet (mmol/L. Mean±SD.n=9).

GroupTCTGLDL-CHDL-CControl5.66±0.610.97±0.182.82±0.080.58±0.08Ator5.74±0.600.97±0.162.87±0.110.58±0.07KO5.52±0.520.98±0.592.90±0.110.57±0.02KO+Ator5.59±0.710.97±0.102.89±0.120.57±0.05Tg5.96±0.540.97±0.072.86±0.120.57±0.03Tg+Ator5.81±0.530.98±0.072.98±0.140.58±0.03

Figure 3. The blood lipid levels of the mice after treatment with high-fat diet and atorvastatin (Ator). Mean±SD.n=9.*P<0.05vscontrol group;#P<0.05vsAtor group;△P<0.05vsKO group;▲P<0.05vsKO+Ator group;$P<0.05vsTg group.
图3各组小鼠干预结束时血脂水平的比较

Figure 4. The total preparation of the mouse aorta with oil red O staining (A) and the quantitative analysis for detecting the relative area of aortic plaque (B). Mean±SD.n=3.*P<0.05vscontrol group;△P<0.05vsKO group;$P<0.05vsTg group.
图4主动脉整体油红O染色和主动脉斑块相对面积的定量分析
4.2主动脉弓HE染色 干预结束后小鼠主动脉弓行HE染色结果可见,对照组、KO组及Tg组镜下见粥样斑块形成,斑块表面为厚薄不均匀的纤维帽,纤维帽下含有泡沫细胞、针状空隙的胆固醇结晶及大量坏死产物,斑块下内弹性膜破坏,可见SMC穿过内弹力膜,中膜SMC及纤维层排列紊乱;其中Tg组粥样斑块较对照组及KO组减少。经Ator干预8周后,Ator组、KO+Ator组及Tg+Ator组镜下见向管腔凸起的斑块形成,斑块表面为纤维帽,纤维帽下见大量泡沫细胞和炎症细胞,未见明显胆固醇空隙和钙盐沉积,斑块下内弹性膜尚完整,中膜可见排列尚整齐的梭形平滑肌细胞,纤维层结构尚完整;其中可见Tg+Ator组粥样斑块较Ator组及KO+Ator组减少,见图5。
4.3主动脉根部HE染色 干预结束后,对小鼠主动脉根部(经瓣膜处)行HE染色,各组均可见斑块形成,但斑块形态及相对面积不一。对照组、KO组及Tg组斑块形成多,斑块内见大量胆固醇结晶、泡沫细胞、坏死产物及炎症细胞;Ator组、KO+Ator组及Tg+Ator组主动脉窦部可见大量斑块形成,斑块内可见大量泡沫细胞及炎症细胞,见少量胆固醇结晶,见图6A。主动脉根部斑块相对面积结果示,Tg组斑块面积少于KO组和对照组(P<0.05),KO组斑块面积多于对照组(P<0.05);经Ator干预8周后,主动脉根部斑块相对面积示Tg+Ator组少于Tg组(P<0.05),KO+Ator组少于KO组(P<0.05),Ator组少于对照组(P<0.05),见图6B。

Figure 5. Observation of the aortic arch with HE staining.
图5主动脉弓的HE染色观察

Figure 6. The observation of aortic root with HE staining (A) and the quantitative analysis for detecting the relative area of mouse aortic root plaque (B). Mean±SD.n=3.*P<0.05vscontrol group;△P<0.05vsKO group;$P<0.05vsTg group.
图6主动脉根部HE染色观察和主动脉根部斑块相对面积的定量分析
5 HSP22、NF-κB、eNOS、ICAM-1及IL-6的检测
5.1主动脉HSP22蛋白表达的变化 Western blot法检测小鼠主动脉HSP22蛋白表达,结果显示Tg组的HSP22蛋白表达高于KO组及对照组(P<0.05),对照组高于KO组(P<0.05)。经Ator干预8周之后,主动脉HSP22表达在KO组与KO+Ator组比较,差异无统计学显著性;Ator组低于对照组(P<0.05),Tg+Ator组低于Tg组(P<0.05);其中Tg+Ator组高于Ator组及KO+Ator组(P<0.05),见图7。免疫组化法检测小鼠主动脉的HSP22蛋白表达,结果显示Tg组及对照组可见阳性着色多,与KO组及Ator组相比HSP22蛋白表达升高(P<0.05);其中对照组高于KO组(P<0.05);经Ator干预8周之后,Tg+Ator组较Tg组阳性着色浅(P<0.05),Ator组较对照组阳性着色浅(P<0.05),KO+Ator组与KO组阳性着色无明显变化;其中Tg+Ator组较Ator组及KO+Ator组阳性着色深,表示HSP22表达较Ator组及KO+Ator组多(P<0.05),见图7B。

Figure 7. The protein expression of HSP22 in the aorta determined by Western blot (A) and immunohistochemistry (B, ×200). Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsAtor group;△P<0.05vsKO group;▲P<0.05vsKO+Ator group;$P<0.05vsTg group.
图7Westernblot法和免疫组化法检测主动脉HSP22蛋白的表达
5.2主动脉NF-κB蛋白表达的变化 Western blot法检测小鼠主动脉NF-κB蛋白的表达,结果显示小鼠主动脉NF-κB蛋白表达在对照组中较KO组降低(P<0.05)、较Tg组升高(P<0.05);经Ator干预8周后,主动脉NF-κB蛋白表达示Ator组较对照组降低(P<0.05),KO+Ator组较KO组降低(P<0.05),Tg+Ator组较Tg组降低(P<0.05);其中KO+Ator组较Ator组及Tg+Ator组升高(P<0.05),而Ator组与Tg+Ator组比较差异无统计学显著性,见图8A。免疫组化法检测小鼠主动脉NF-κB蛋白表达,结果可见对照组、KO组及Tg组小鼠主动脉棕色着色深,其中,KO组棕色着色深于对照组及Tg组,表示NF-κB蛋白表达高于Tg组(P<0.05),其中Tg组低于对照组(P<0.05)。经Ator干预8周后,Ator组较对照组棕色着色浅(P<0.05),KO+Ator组较KO组棕色着色浅(P<0.05),Tg+Ator组较Tg组棕色着色浅(P<0.05);而Ator组、KO组与Tg+Ator之间差异无统计学显著性,见图8B。
5.3主动脉eNOS蛋白表达的变化 Western blot法检测小鼠主动脉eNOS蛋白表达的结果显示,Tg组及对照组较KO组升高(P<0.05)。经Ator干预8周后,小鼠主动脉eNOS蛋白的表达Ator组较对照组升高(P<0.05),KO+Ator组较KO组升高(P<0.05),Tg+Ator组较Tg组升高(P<0.05);其中KO+Ator组较Ator组及Tg+Ator组下降(P<0.05),见图9。
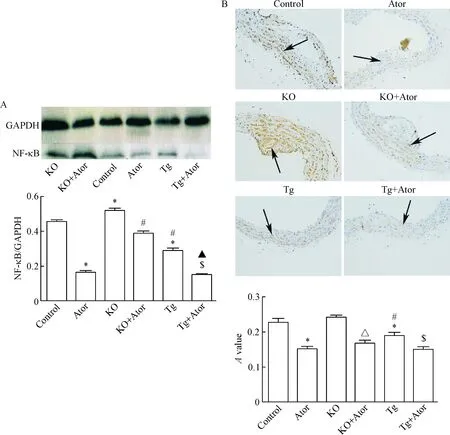
Figure 8. The protein expression of NF-κB in the aorta determined by Western blot (A) and immunohistochemistry (B, ×200). Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsAtor group;△P<0.05vsKO group;▲P<0.05vsKO+Ator group;$P<0.05vsTg group.
图8Westernblot法和免疫组化法检测主动脉NF-κB蛋白的表达
5.4主动脉ICAM-1蛋白表达的变化 Western blot法检测小鼠主动脉ICAM-1蛋白表达的结果显示,KO组高于对照组及Tg组(P<0.05),其中Tg组低于对照组(P<0.05)。经Ator干预8周后,小鼠主动脉ICAM-1蛋白的表达示Ator组低于对照组(P<0.05),KO+Ator组较KO组降低(P<0.05),Tg+Ator组较Tg组降低(P<0.05);其中KO+Ator组较Ator组及Tg+Ator组升高(P<0.05),Tg+Ator组较Ator组降低(P<0.05),见图10。
5.5血清IL-6浓度的变化 ELISA检测小鼠血清IL-6的浓度,结果显示对照组、KO组及Tg组小鼠血清IL-6水平的差异无统计学显著性。在Ator干预8周之后,血清中IL-6浓度在Ator组低于对照组(P<0.05),KO+Ator组低于KO组(P<0.05), Tg+Ator组低于Tg组(P<0.05);而血清IL-6水平在Ator组、KO+Ator组及Tg+Ator组间比较,差异无统计学显著性,见图11A。
5.6血清HSP22浓度的变化 ELISA检测小鼠血清HSP22的浓度,结果显示KO组低于对照组和Tg组(P<0.05),对照组低于Tg组(P<0.05)。经Ator干预8周后,小鼠血清HSP22浓度在Ator组低于对照组(P<0.05),KO+Ator组与KO组比较,差异无统计学显著性,Tg+Ator组低于Tg组(P<0.05),KO+Ator组低于Ator组及Tg+Ator组(P<0.05),Tg+Ator组高于Ator组(P<0.05),见图11B。
讨 论
1 动脉粥样硬化
AS的危险因素包括高脂血症、高血压、高血糖、吸烟、血管炎症及血栓形成等。这些因素的存在会导致机体的活性氧簇的产生,炎症因子表达增加,其中尤其是脂质的异常,机体ROS会将LDL氧化为ox-LDL,而ox-LDL会促使内皮细胞渗透性增加,激活单核细胞迁移到血管壁,上调黏附分子及炎症因子的表达,而迁移到血管壁的单核细胞分化为巨噬细胞,ox-LDL被巨噬细胞所摄取形成泡沫细胞,沉积在受损的内皮细胞表面[14]。这些变化进一步导致内皮细胞损伤及炎症反应,加速AS的进展,因此抗炎、抗氧化应激是治疗AS的重要途径。
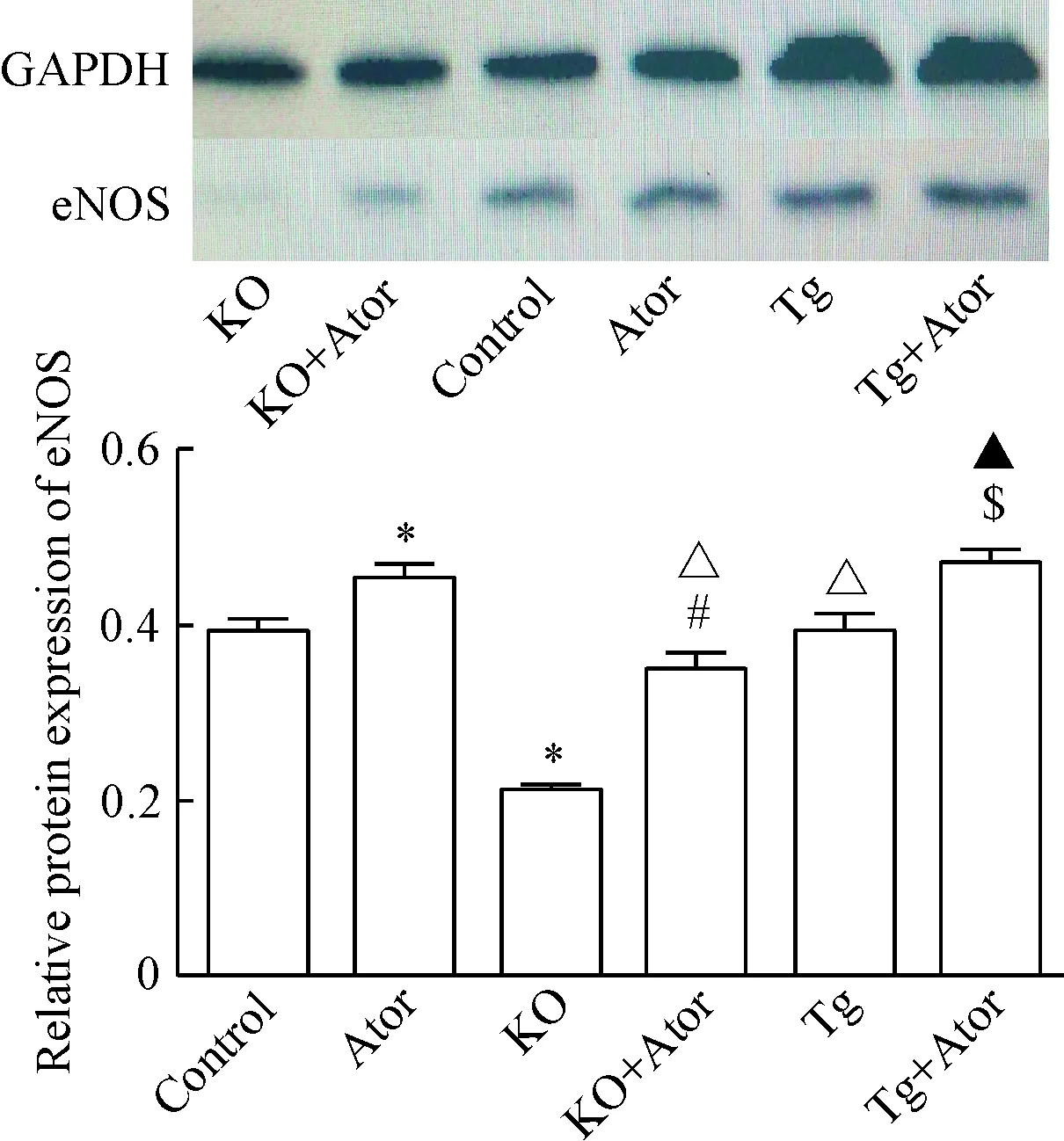
Figure 9. The protein expression of eNOS in the aorta determined by Western blot. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsAtor group;△P<0.05vsKO group;▲P<0.05vsKO+Ator group;$P<0.05vsTg group.
图9Westernblot法检测主动脉eNOS蛋白的表达

Figure 10. The protein expression of ICAM-1 in the aorta determined by Western blot. Mean±SD.n=3.*P<0.05vscontrol group;#P<0.05vsAtor group;△P<0.05vsKO group;▲P<0.05vsKO+Ator group;$P<0.05vsTg group.
图10Westernblot法检测主动脉ICAM-1蛋白的表达
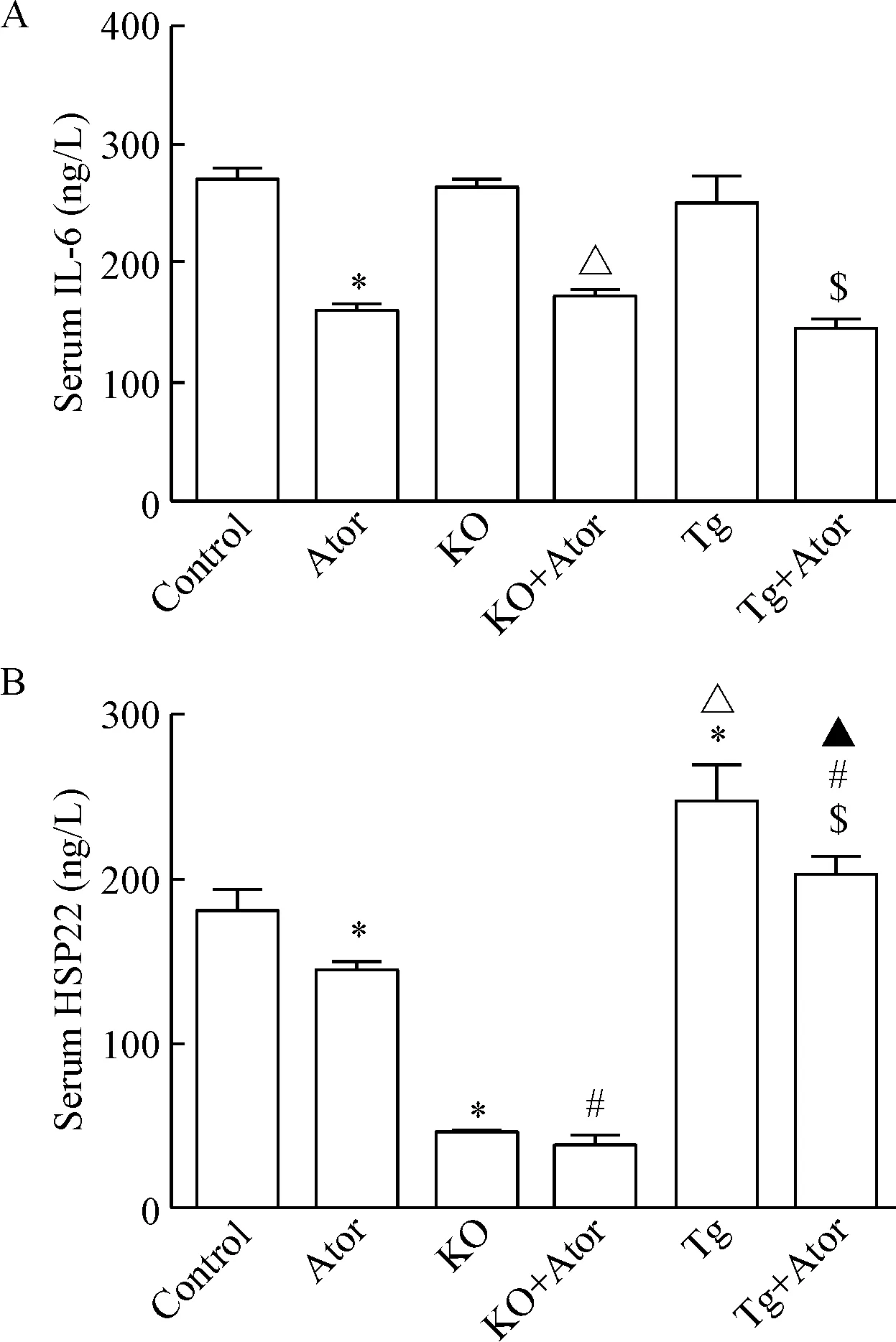
Figure 11. The concentrations of serum IL-6 (A) and HSP22 (B) were measured by ELISA. Mean±SD.n=6.*P<0.05vscontrol group;#P<0.05vsAtor group;△P<0.05vsKO group;▲P<0.05vsKO+Ator group;$P<0.05vsTg group.
图11ELISA检测血清IL-6和HSP22的浓度
我们前期研究已经成功构建小鼠AS模型,将此方法应用于本研究中[15],通过对所有小鼠进行高脂饲料喂养12周构建AS模型,结果发现,对照组和KO组小鼠体重、血脂及炎症因子IL-6、ICAM-1和NF-κB的水平分别明显高于Ator组及KO+Ator组,主动脉根部及弓部肉眼和体视镜下观察发现斑块较多,镜下见斑块内大量泡沫细胞形成,血管平滑肌细胞向管壁内膜迁移,斑块内胆固醇结晶形成,由此说明,本研究成功构建了小鼠AS模型。
在本研究中,各组小鼠基础体重及血脂水平无明显差异,在高脂饮食12周后,小鼠血清中TG及HDL-C的含量在对照组、KO组及Tg组间的差异无统计学显著性,Tg组血清的LDL-C及TC较KO组显著降低,说明HSP22可直接降低LDL-C水平,其作用可能是通过降低炎症反应来发挥的,具体机制还需进一步验证。他汀干预8周后,Ator组的血清TC及LDL-C低于对照组、HDL-C高于对照组,KO+Ator组血清TC及LDL-C低于KO组、HDL-C高于KO组,Tg+Ator组血清TC及LDL-C低于Tg组、HDL-C高于Tg组;Ator组、KO+Ator组及Tg+Ator组血清LDL-C及HDL-C间的差异无统计学显著性,由此说明他汀的调脂作用远远强于HSP22;各组小鼠血清中的TG在干预前后无显著变化,说明他汀及HSP22对TG无明显影响。就小鼠主动脉根部斑块面积而言,Tg组主动脉斑块相对面积低于KO组,说明HSP22过表达可改善AS的进展,他汀干预8周后,主动脉斑块面积于Ator组低于对照组,KO+Ator组低于KO组,Tg+Ator组低于Tg组;其中Tg+Ator组主动脉斑块面积较KO+Ator组减少,说明HSP22可促进他汀进一步改善AS。综合以上结果,说明HSP22基因缺失及过表达会影响AS进展,HSP22基因缺失会加重AS进展,过表达可发挥抗AS作用,他汀具有显著抗AS作用,HSP22与他汀具有协同作用,可促进他汀调脂及改善AS作用。
2 HSP22在AS中的表达及作用
在缺氧状态下,心肌细胞胞质及线粒体内HSP22能上调一氧化氮合酶(nitric oxide synthase,NOS)的表达,增加NO产生,调节线粒体呼吸功能,减少氧化磷酸化及ROS的产生及线粒体转换孔的开放,起到保护心肌细胞效应[16]。本团队前期研究发现,高脂血症致大鼠AS模型中,主动脉HSP22蛋白表达显著升高,他汀治疗后可降低HSP22蛋白的表达,缓解AS进展[13]。用ox-LDL处理人脐静脉内皮细胞可引起HSP22蛋白表达升高,而干扰HSP22表达时会加重ox-LDL所致的细胞凋亡,表明HSP22可改善ox-LDL诱导的细胞损伤,对细胞起到保护作用[12]。
本研究用Western blot和免疫组化法检测小鼠主动脉的HSP22表达,发现KO组及KO+Ator组间的差异无统计学显著性,Tg组高于KO组及对照组,对照组高于KO组,这与主动脉斑块面积、炎症因子表达的变化一致,由此说明长期高脂饮食诱导AS,在此内皮损伤刺激下,HSP22应激性表达升高,从而对血管损伤产生保护效应,而HSP22过表达在AS发生发展中起到抵抗应激的作用,从而改善AS。HSP22可定位于细胞内,也可旁分泌到细胞外,前期研究发现高脂血症小鼠较对照组,血清HSP22的浓度显著增加,考虑是因为长期慢性炎症刺激导致细胞应激性反应使HSP22的表达上调,并可向细胞外分泌(如血液)。在本实验中,发现小鼠血清中HSP22浓度在KO组及KO+Ator组未见明显差异,Tg组高于KO组及对照组,对照组高于KO组,这与主动脉HSP22蛋白表达情况一致,由此说明胞内外HSP22水平变化是一致的。这是一个观察性研究,未深入探讨胞外(如血清)HSP22浓度增加的机制。胞外HSP22是否具有胞内HSP22的抗炎、抗氧化应激、抗凋亡等作用,目前尚未有研究报道。
3 HSP22对阿托伐他汀干预的影响
他汀类药物如阿托伐他汀是HMG-CoA还原酶抑制剂,除了具有调脂作用外,还有改善内皮功能、抗炎、抗氧化应激、稳定斑块及抑制血栓形成等作用。他汀的降脂治疗是基于降低LDL水平,且部分是通过升高HSP90表达从而上调eNOS并可稳定eNOS的mRNA表达而起作用的[17]。
前期研究结果显示用他汀治疗AS模型大鼠,可降低HSP22蛋白的表达,缓解AS进展。在本研究中发现,在他汀干预8周之后,KO组与KO+Ator组主动脉及胞外血清中HSP22无显著差异,而Ator组及Tg+Ator组主动脉及胞外血清中HSP22表达较相应对照组显著降低,与主动脉斑块面积、炎症因子IL-6、ICAM-1、NF-κB以及血脂的变化相一致,并与前期研究结果一致。由此说明他汀可降低HSP22表达,提示他汀未能通过上调HSP22的表达来改善AS进展。主动脉及血清中HSP22蛋白表达在Tg+Ator组中高于Ator组及KO+Ator组。而斑块面积及炎症因子则相反,由此推测,HSP22过表达与他汀具有协同作用,可进一步放大他汀的抗炎及调脂效应,进一步改善AS,而AS改善后,应激刺激减少,主动脉及血清中HSP22表达反馈性下降。
4 eNOS与HSP22的关系及他汀干预的影响
内皮损伤是AS病变的起始因素,内皮功能在很大程度上依赖eNOS的表达,由eNOS合成的NO能抑制内皮细胞黏附分子表达、细胞迁移、增殖,并调节血管张力及血小板激活等,高脂血症可致细胞eNOS合成的NO减少,NO的生物利用度降低[18]。eNOS催化L-精氨酸和分子氧生成L-瓜氨酸和NO,NO不仅是一种血管舒张因子,还具有调节平滑肌细胞的迁移、白细胞的黏附及抗血小板聚集的作用,影响着心血管系统疾病的生理病理过程[19]。在本团队前期研究中发现,高脂饮食致大鼠AS模型中主动脉HSP22表达水平升高,eNOS水平下降,他汀治疗后可降低主动脉HSP22蛋白的表达,升高eNOS水平,缓解AS进展[13],在细胞层面上,HSP22作用于HUVECs,发现eNOS和p-eNOS上调,促进了NO的释放,内皮细胞功能得到改善[12]。
在本研究中,所有小鼠经12周高脂饮食后,主动脉eNOS蛋白表达在Tg组和对照组高于KO组,说明在高脂应激下HSP22可上调eNOS表达。他汀治疗8周后,主动脉的eNOS蛋白表达在Ator组较对照组升高,KO+Ator组较KO组升高,Tg+Ator组较Tg组升高,说明他汀干预可上调eNOS表达;其中KO+Ator组eNOS的蛋白表达较Ator组及Tg+Ator组下降。由此可见HSP22基因缺失部分限制了他汀上调eNOS表达的作用,但过表达并没有促进他汀上调eNOS表达。他汀对eNOS的调控作用远远强于HSP22,两者可能是通过不同信号通路对eNOS表达进行调节。
5 NF-κB与HSP22的关系及他汀干预的影响
HSP90可以抑制NF-κB的产生和信号转导及转录激活因子的活性,从而降低糖尿病小鼠糖尿病肾病及心血管系统疾病的发生率[20]。在本研究中,经12周高脂饮食后,小鼠主动脉NF-κB蛋白表达在对照组低于KO组、高于Tg组,由此说明HSP22参与了NF-κB表达的调控,过表达HSP22可抑制NF-κB的激活;他汀治疗8周之后,主动脉NF-κB蛋白表达在Ator组较对照组降低,KO+Ator组较KO组降低,Tg+Ator组较Tg组降低;其中KO+Ator组较Ator组及Tg+Ator组升高;说明他汀干预可降低NF-κB表达,HSP22基因缺失部分限制了他汀下调NF-κB表达的作用,但过表达并没有促进他汀下调NF-κB表达的作用,且他汀对NF-κB表达的调控作用远强于HSP22,两者可能是通过不同的信号通路对NF-κB 进行调控。
6 ICAM-1与HSP22的关系及他汀干预的影响
胆固醇作用于人内皮细胞可通过盐皮质激素受体调节ICAM-1的转录水平。对ApoE-/-小鼠喂养胆固醇4周后,ICAM-1表达升高,这与主动脉根部斑块面积、脂质水平及炎症因子增加是一致的;在ICAM-1与ApoE双敲除(ApoE-/-/ICAM-1-/-)小鼠AS模型中,斑块面积、脂质水平及巨噬细胞渗透性下降了[21]。在应激状态下,线粒体中的HSP60会迁移到细胞质内,并协同黏附分子(ICAM-1、VCAM-1、ELAM-1)大量表达于细胞表面,诱导炎症反应,另外,在感染、机械应激等内环境稳态改变时,HSP60本身也会诱导内皮细胞产生VCAM-1、ICAM-1及IL-6的表达,这些分子的存在加速了AS的进展[22]。
在本研究中,高脂饮食12周后,小鼠主动脉ICAM-1蛋白表达在KO组高于对照组及Tg组,其中Tg组低于对照组,由此说明HSP22可降低ICAM-1的表达;经Ator干预8周后,小鼠主动脉ICAM-1蛋白表达在Ator组低于对照组,KO+Ator组低于KO组,Tg+Ator组低于Tg组;其中KO+Ator组低于Ator组及Tg+Ator组,Tg+Ator组低于Ator组;说明他汀可降低ICAM-1的表达。HSP22基因缺失部分抑制他汀降ICAM-1表达的作用,过表达可促进他汀降ICAM-1表达,两者具有协同抗炎作用,进一步改善AS进展。
7 IL-6与HSP22的关系及他汀干预的影响
研究者用ELISA法定量检测发现,AS病变区域中的IL-6和IL-8水平显著高于正常内膜;内皮细胞功能测定显示,AS病变区域内皮活化程度亦显著高于正常内膜,表明IL-6及IL-8可能参与了AS早期炎症及内皮活化过程[23]。在本研究中,高脂饮食12周后,小鼠血清IL-6浓度在对照组、KO组及Tg组间的差异无统计学显著性,说明HSP22对 IL-6释放的调节作用不明显;他汀干预8周后,小鼠血清IL-6在Ator组低于对照组,KO+Ator组低于KO组, Tg+Ator组低于Tg组,其中Ator组、KO+Ator组及Tg+Ator组血清IL-6水平的差异无统计学显著性,说明他汀干预可显著降低血清中IL-6释放,而HSP22可能不参与此过程。
本文结果提示,HSP22基因缺失可上调NF-κB和ICAM-1的表达,降低eNOS的表达,加速AS的进展,HSP22基因过表达可降低NF-κB和ICAM-1的表达,升高eNOS的表达,从而改善AS。高脂饮食上调NF-κB、ICAM-1和IL-6的表达,下调eNOS表达,诱导AS形成,他汀可上调eNOS的表达,下调NF-κB、IL-6和ICAM-1的表达,改善AS。HSP22基因缺失部分限制了他汀下调NF-κB及ICAM-1、上调eNOS表达的作用,其过表达可促进他汀下调ICAM-1表达,进一步改善AS。但因本实验为观察性研究,主动脉HSP22蛋白作用的具体机制仍需进一步验证。对汀干预会影响胞外血清中HSP22浓度这一现象,后续研究应深入探讨血清HSP22浓度变化的机制及其对血管AS病变的影响。
[参 考 文 献]
[1] Ketelhuth DF, Hansson GK. Adaptive response of T and B cells in atherosclerosis[J]. Circ Res, 2016, 118(4):668-678.
[2] Mitra S, Deshmukh A, Sachdeva R, et al. Oxidized low-density lipoprotein and atherosclerosis implications in antioxidant therapy[J]. Am J Med Sci, 2011, 342(2):135-142.
[3] Anogeianaki A, Angelucci D, Cianchetti E, et al. Atherosclerosis: a classic inflammatory disease[J]. Int J Immunopathol Pharmacol, 2011, 24(4):817-825.
[4] Acunzo J, Katsogiannou M, Rocchi P. Small heat shock proteins HSP27 (HspB1), αB-crystallin (HspB5) and HSP22 (HspB8) as regulators of cell death[J]. Int J Biochem Cell Biol, 2012, 44(10):1622-1631.
[5] Cuerrier CM, Chen YX, Tremblay D, et al. Chronic over-expression of heat shock protein 27 attenuates atherogenesis and enhances plaque remodeling: a combined histological and mechanical assessment of aortic lesions[J]. PLoS One, 2013, 8(2):e55867.
[6] Almanzar G, Ollinger R, Leuenberger J, et al. Autoreactive HSP60 epitope-specific T-cells in early human atherosclerotic lesions[J]. J Autoimmunity, 2012, 39(4):441-450.
[7] Han WQ, Chang FJ, Wang QR, et al. Microparticles from patients with the acute coronary syndrome impair vasodilatation by inhibiting the Akt/eNOS-Hsp90 signaling pathway[J]. Cardiology, 2015, 132(4):252-260.
[8] Wendling U, Paul L, van der Zee R, et al. A conserved mycobacterial heat shock protein (hsp) 70 sequence prevents adjuvant arthritis upon nasal administration and induces IL-10-producing T cells that cross-react with the mammalian self-hsp70 homologue[J]. J Immunol, 2000, 164(5):2711-2717.
[9] Xie F, Zhan R, Yan LC, et al. Diet-induced elevation of circulating HSP70 may trigger cell adhesion and promote the development of atherosclerosis in rats[J]. Cell Stress Chaperones, 2016, 21(5):907-914.
[10] Bakthisaran R, Tangirala R, Rao ChM. Small heat shock proteins: role in cellular functions and pathology[J]. Biochim Biophys Acta, 2015, 1854(4):291-319.
[11] Marunouchi T, Abe Y, Murata M, et al. Changes in small heat shock proteins HSPB1, HSPB5 and HSPB8 in mitochondria of the failing heart following myocardial infarction in rats[J]. Biol Pharm Bull, 2013, 36(4):529-539.
[12] 叶燕平. 在ox-LDL致内皮细胞损伤中HSP22的保护作用及他汀的干预作用 [D]. 南昌: 南昌大学, 2014.
[13] 方海洋, 陈 琦, 吴延庆, 等. 高脂血症大鼠主动脉HSP22、TNF-α和eNOS的表达及阿托伐他汀的影响[J]. 中国病理生理杂志, 2014, 30(10):1873-1878.
[14] Zhang M, Jiang L. Oxidized low-density lipoprotein decreases VEGFR2 expression in HUVECs and impairs angiogenesis[J]. Exp Ther Med, 2016, 12(6):3742-3748.
[15] 涂小丽, 陈 琦, 吴延庆, 等. CXCR7在动脉粥样硬化模型ApoE-/-小鼠的表达及阿托伐他汀的干预作用[J]. 中国病理生理杂志, 2015, 31(12):2209-2215.
[16] Laure L, Long R, Lizano P, et al. Cardiac H11 kinase/Hsp22 stimulates oxidative phosphorylation and modulates mitochondrial reactive oxygen species production: involvement of a nitric oxide-dependent mechanism[J]. Free Radical Biol Med, 2012, 52(11-12):2168-2176.
[17] Brouet A, Sonveaux P, Dessy C, et al. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins[J]. Circ Res, 2001, 89(10):866-873.
[18] Vergnani L, Hatrik S, Ricci F, et al. Effect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production: key role of L-arginine availability[J]. Circulation, 2000, 101(11):1261-1266.
[19] Siragusa M, Frohlich F, Park EJ, et al. Stromal cell-derived factor 2 is critical for Hsp90-dependent eNOS activation[J]. Sci Signal, 2015, 8(390):ra81.
[20] Jo HS, Kim DW, Shin MJ, et al. Tat-HSP22 inhibits oxidative stress-induced hippocampal neuronal cell death by regulation of the mitochondrial pathway[J]. Mol Brain, 2017, 10:1.
[21] Marzolla V, Armani A, Mammi C, et al. Essential role of ICAM-1 in aldosterone-induced atherosclerosis[J]. Int J Cardiol, 2017, 232:233-242.
[22] Kilic A, Mandal K. Heat shock proteins: pathogenic role in atherosclerosis and potential therapeutic implications[J]. Autoimmune Dis, 2012, 2012:502813.
[23] Rus HG, Vlaicu R, Niculescu F. Interleukin-6 and interleukin-8 protein and gene expression in human arterial atherosclerotic wall[J]. Atherosclerosis, 1996, 127(2):263-271.
