玉米种子萌发相关性状的全基因组关联分析
2018-05-14田润苗张雪海汤继华白光红付志远
田润苗 张雪海 汤继华 白光红 付志远,*
玉米种子萌发相关性状的全基因组关联分析
田润苗1,**张雪海1,**汤继华1白光红2付志远1,*
1河南农业大学农学院, 河南郑州 450002;2新疆农业大学, 新疆乌鲁木齐 830052
种子萌发是出苗的前提, 对玉米产量影响重大。为了解玉米种子萌发相关性状的遗传机制, 本研究对476份玉米自交系种子萌发相关的6个性状进行调查, 结合125万个(1.25M) SNP标记, 利用3种统计模型(Q, K, Q+K)进行全基因关联分析(GWAS)。结果表明K模型能够较好地评价吸胀前重量、吸胀前体积、吸胀后重量、吸胀后体积和吸胀体积5个性状; Q+K模型能更好地评价吸胀重量性状。基于这6个性状的最优模型的GWAS结果, 共检测到15个种子萌发相关性状的显著SNP, 15个SNP对应6个QTL, 集中分布在玉米第3、第6、第7和第10染色体上, QTL内单个SNP能解释的表型变异为5.09%~7.85%。其中5个QTL可在多个生物学重复中被检测到。以最显著SNP所在基因或附近基因作为QTL的候选基因, 共筛选到6个最可能的候选基因。GRMZM2G148411是吸胀后重量、吸胀重量和吸胀体积3个性状共同鉴定到的QTL候选基因, 根据基因的功能注释, 该基因编码一个包含TLD-domain的钙离子结合蛋白, 可能是一种调控种子休眠与萌发的信号分子。本研究鉴定的QTL为解析玉米种子萌发的遗传机制和相应功能标记的开发奠定了基础。
玉米; 种子萌发; 全基因组关联分析; 候选基因
随着玉米机械化收获程度的提高, 生产上对玉米种子田间出苗率和出苗整齐度的要求越来越高。种子的高效萌发是种子出苗的前提[1]。大量生产实践证明不同玉米自交系的发芽率和出苗率均存在广泛的遗传变异[2-3], 解析玉米种子出苗能力的遗传基础, 发掘玉米萌发相关性状的优良等位基因, 对玉米产量的提高及农业机械化收获具有重要意义。
全基因组关联分析(genome-wide association studies, GWAS)是研究数量性状的重要方法之一, 最早用于人类疾病研究[4]。近年来, 随着新一代测序技术的发展, GWAS在植物重要性状遗传机制研究中的应用越来越多。Atwell等[5]在拟南芥中首次证明应用GWAS的可行性, 并指出此方法也适用于其他植物。玉米具有丰富的遗传变异和快速的连锁不平衡衰减, 是遗传学研究的重要模式作物。近十年来, 以玉米为材料的GWAS取得了很大进展[7]。以本研究所用关联群体为例, Li等[8]用1.03万个SNP标记对籽粒含油量进行GWAS, 共检测出74个显著位点。Liu等[9]通过整合简化基因组测序、高密度SNP芯片及深度RNA测序数据, 开发一套包含1.25M SNP的标记(MAF≥AFP), 并以此重新检测了关联群体的籽粒含油量, 检测到13个新的显著位点。而利用GWAS对种子萌发相关性状进行遗传分析的报道相对较少。Huang等[10]用5.6万个SNP标记对125个玉米自交系在种子萌发阶段和幼苗期的耐冷性进行GWAS, 共检测到43个显著位点。Shi等[11]利用GWAS对水稻种子在盐胁迫条件下的7个萌发相关性状进行分析, 检测到11个主效位点。Kan等[12]用大豆种子萌发阶段耐盐性的GWAS检测到8个显著SNP。
种子出苗受多种因素影响, 如种子萌发能力、种子活力等。种子萌发包含吸胀和代谢过程的起始、吸水滞后期、进一步吸水后胚根出现3个阶段[13]。目前的研究主要集中在种子萌发的第二和第三阶段。Fu等[14]利用蛋白质组学方法获得了玉米种子萌发第二阶段的杂种优势相关差异表达蛋白。Hu等[15]对玉米种子低温条件下萌发能力的QTL分析, 鉴定出6个种子发芽率的QTL及6个胚根长度的QTL。Han等[16]在人工老化的条件下对玉米种子的发芽率、发芽势、幼苗干重及幼苗根部干重的QTL分析, 共检测到65个QTL, 结合代谢组mQTL分析, 共鉴定出23个候选基因, 这些基因通过调控糖酵解通路和蛋白质代谢进而影响种子的萌发。种子吸胀作为种子休眠结束和种子萌发第一阶段, 对种子的正常萌发具有重要意义, 但相关的遗传研究很少。Farzaneh等[17]利用反向遗传学方法找到一些在拟南芥种子吸胀过程中差异表达的基因, 这些基因的缺失突变体表现出与种子萌发和休眠相关的表型。Li等[18]研究水稻家族基因时, 发现6个与种子吸胀有关的基因。Noblet等[19]在玉米种子低温吸胀过程中的细胞膜磷脂重组的研究中发现, 玉米种子中饱和脂肪酸与不饱和脂肪酸的差异积累与种子吸胀过程中对低温的敏感性有关。目前有关玉米种子吸胀相关性状的QTL研究尚无报道。为了解玉米种子吸胀相关性状的遗传机制, 本研究对476份玉米自交系种子吸胀前后及吸胀程度的6个相关性状进行测定, 利用1.25M的SNP标记(http://www. maizego.org/Resources.html)对这些性状进行GWAS, 以挖掘影响种子萌发的相关候选基因, 并为相应基因的克隆及功能标记的开发奠定基础。
1 材料与方法
1.1 试验设计与性状调查
476份玉米自交系来自温带、热带及亚热带(http://www.maizego.org/Resources.html), 于2017年春季在室内进行发芽实验, 共3个生物学重复, 每个生物学重复3次抽样测定, 每次抽样均由20粒大小一致的健康种子构成。种子浸泡前, 分别测定每次抽样的20粒干种子的重量(W1)和排水体积(V1); 种子吸胀24 h后(温度26℃, 湿度60%), 用滤纸拭干种子表面的水分, 再次测定每次抽样的重量(W2)和排水体积(V2); 每次抽样的吸胀重量W3和吸胀体积V3分别由W3=W2–W1和V3=V2–V1公式计算得到。以每个生物学重复3次抽样的平均值作为该重复的值, 以每个材料3个生物学重复的平均值作为该性状的值。对每个生物学重复及其平均值分别进行GWAS。测定完种子性状后, 继续培养(室内发芽盒中土培)以确保种子萌发出苗。
1.2 统计分析
使用SPSS 23.0软件对各表型数据进行双因素方差分析(two-way ANOVA)、皮尔逊相关性分析(Pearson correlation analysis)及正态性检验(normality test)。
1.3 全基因组关联分析
1.25M SNP基因型数据从Maizego网站获得(http://www.maizego.org/Resources.html), 在TASSEL3.0软件中, 分别用只控制群体结构的Q模型、只控制亲缘关系的K模型, 以及同时控制群体结构和亲缘关系的Q+K模型实现关联分析。根据Quantile- Quantile 散点图(QQ plot)对每一个性状在3种模型下的结果进行比较, 并选择最优模型下的GWAS结果作为后续QTL分析的基础。每个性状的曼哈顿图和QQ plot均利用R完成(R Core Team 2012, http:// www.R-project.org/)。以=2.04×10–6(1/,为GEC软件计算出的有效标记数490 547)作为显著关联SNP的阈值[20]。
1.4 候选基因的筛选
由于该关联群体10条染色体的平均连锁不平衡衰减(LD)距离为30 kb[9], 本研究以显著SNP上下游各延伸30 kb的区间定义为一个QTL, 当相邻QTL物理区间有重叠时(B73 v2版本), 则认为是同一个QTL。以每个QTL内最显著SNP所在基因或邻近基因作为该QTL的首要候选基因, 利用MaizeGDB、NCBI及TAIR网站对QTL内的候选基因进行功能注释和分析。
2 结果与分析
2.1 种子萌发相关性状的统计分析
6个性状均表现广泛的遗传变异, 其中, 吸胀前体积(V1)的变异系数最小为17.35%, 吸胀体积的变异系数最大为28.41% (表1)。所有性状的频次分布 (图1)、峰度和偏度(绝对值均小于1)都表现出明显的正态分布特征, 说明这6个性状均是由微效多基因控制的典型数量性状。
方差分析和相关性分析结果表明, 6个性状在不同材料间均存在极显著差异(表2), 且6个性状两两之间均极显著相关(表3), 说明这些性状彼此影响, 且基因型的差异是导致其性状差异的主要原因, 环境因素对性状的表现影响较小。
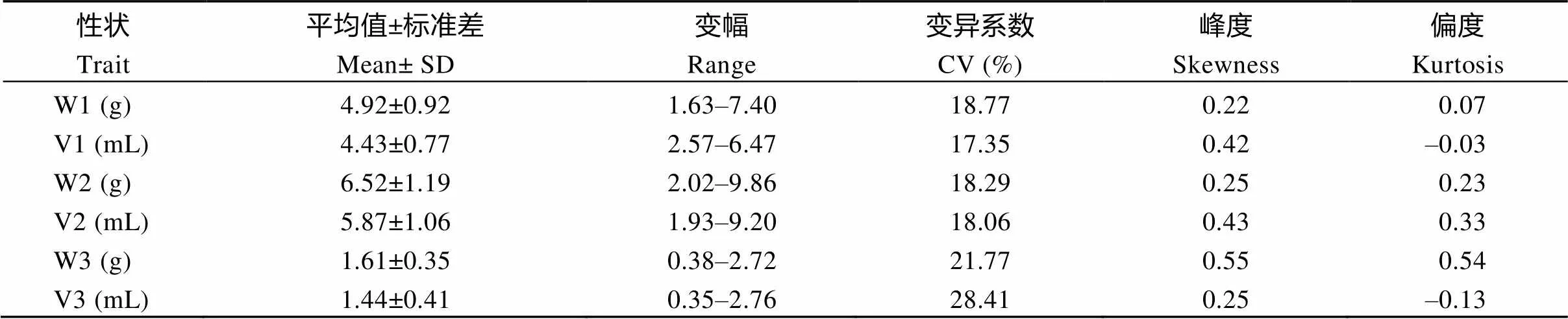
表1 种子萌发性状表型统计分析
W1: 吸胀前重量; V1: 吸胀前体积; W2: 吸胀后重量; V2: 吸胀后体积; W3: 吸胀重量; V3: 吸胀体积。
W1: weight before imbibition; V1: volume before imbibition; W2: weight after imbibition; V2: volume after imbibition; W3: weight of imbibition; V3: volume of imbibition.
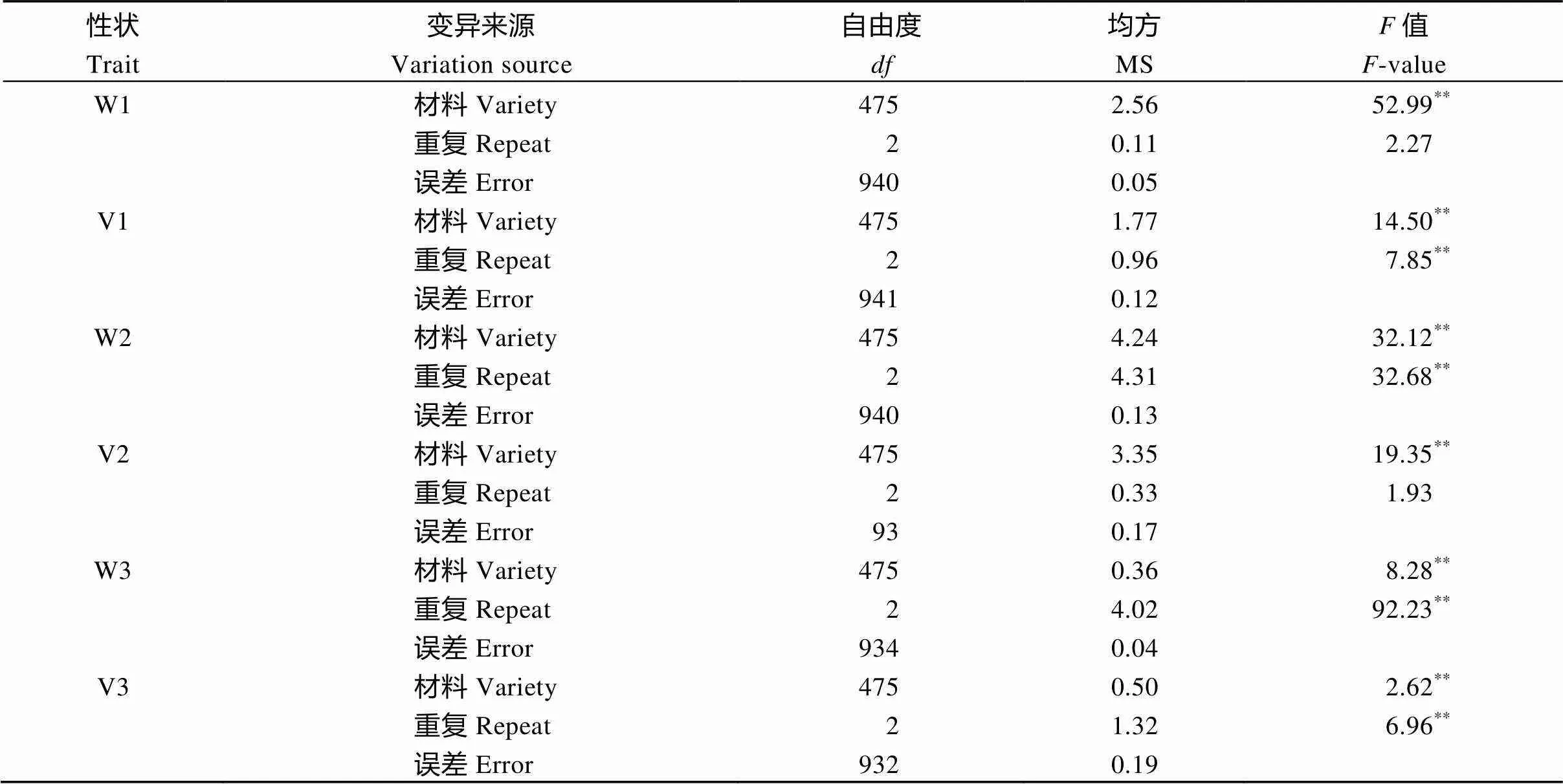
表2 种子萌发性状方差分析
各性状详细名称见表1。*和**分别表示在0.05和0.01水平差异显著。
Name of each trait is given in Table 1.*,**stand for significant at the 0.05 and 0.01 probability levels, respectively
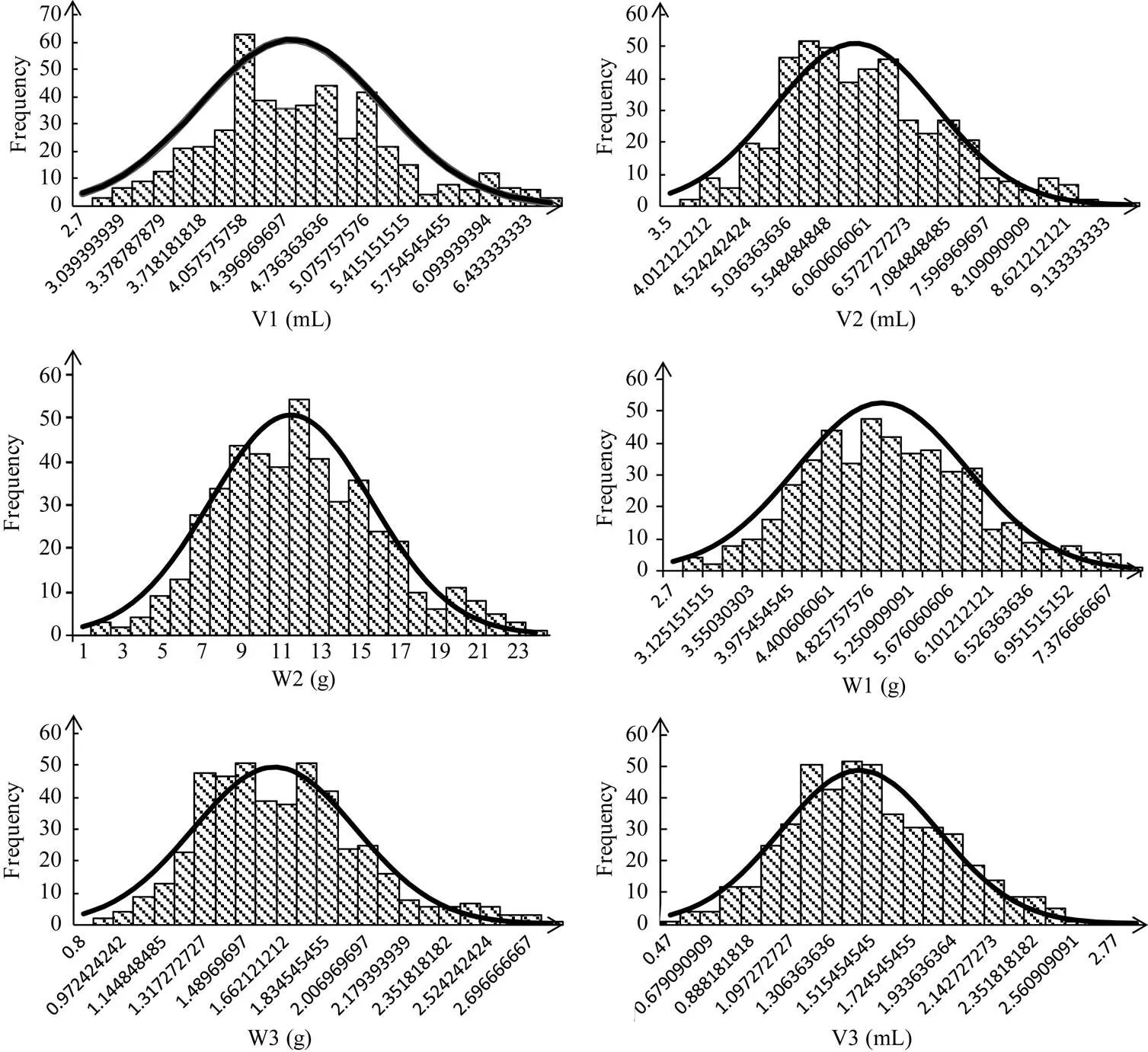
图1 种子萌发性状的频次分布图
各性状详细名称见表1。Name of each trait is given in Table 1.
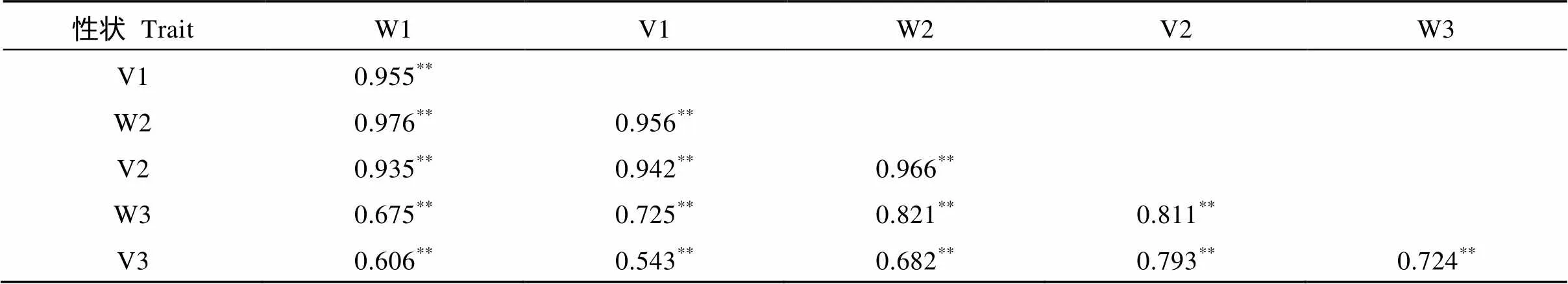
表3 种子萌发性状皮尔逊关联分析
各性状详细名称见表1。*和**分别表示在0.05和0.01水平差异显著。
Name of each trait is given in Table 1.*,**stand for significant at the 0.05 and 0.01 probability levels, respectively.
2.2 关联分析最优模型选择
从3种模型的Q-Q图可以看出(图2), Q模型的假阳性最高(产生更多的I型错误)。对吸胀前重量、吸胀前体积、吸胀后重量、吸胀后体积和吸胀体积性状而言, K模型的拟合效果最好, 即–lg()的真实值最接近预测值, 可以很好地控制假阳性, 因此, 选择K模型的GWAS结果对这5个性状进行QTL解析; 对吸胀重量性状而言, Q+K模型拟合效果最好。因此, 选择Q+K模型作为吸胀重量性状GWAS的最终模型。
2.3 全基因组关联分析与候选基因筛选
基于最优模型, 在阈值为≤2.04×10–6(1/, 1/490 547)时, 共检测到与6个性状显著关联的15个SNP, 分布在玉米第3、第6、第7、第10染色体上(图3和表4)。15个SNP对应6个QTL, 其中, 第7染色体上有2个QTL, 分别影响吸胀前重量(Chr7.S_4828028所在QTL)和吸胀体积(Chr7.S_145970024所在QTL), 可分别解释5.84%和5.13%的表型变异; 第10染色体上的QTL (Chr10.S_6373105所在QTL), 可同时影响吸胀后重量、吸胀重量和吸胀体积, 说明该位点为一因多效位点, 可解释的表型变异为5.17%~6.95%; 第6染色体上检测出2个QTL, 均影响吸胀前体积(Chr6.S_95604386所在QTL和Chr6.S_95604386所在QTL), 可分别解释5.28%和5.93%的表型变异; 第3染色体上检测出1个QTL (Chr3.S_197658474所在QTL), 可解释5.93%的吸涨体积表型变异; 但没有检测到与吸胀后体积显著相关的SNP (图3)。各生物学重复的GWAS结果与各性状均值的GWAS结果高度一致, 除影响吸胀体积的chr3.S_197658474所在QTL外, 其余QTL均在一个或多个生物学重复中被检测到(表5和图4)。

图2 种子萌发性状3种模型比较的QQ图
各性状详细名称见表1。横轴表示经过负的常数对数转换的期望值, 纵轴表示经过负的常数对数转换观察到的值。
Detailed name of each trait was given in Table 1. The horizontal axis shows −lg transformed expected-values, and the vertical axis shows –lg transformed observed-values.
6个QTL内共涉及到44个候选基因(表4), 其中仅有34% (15/44)有功能注释, 根据候选基因的功能注释, 选择最显著SNP所在基因或邻近基因作为首要候选基因(表4)。其中, 吸胀前重量候选基因GRMZM2G167892编码籽粒成熟蛋白(seed mature protein)。同时影响吸胀后重量、吸胀重量和吸胀体积的候选基因GRMZM2G148411 (第10染色体上), 编码一个包含TLD结构域的钙离子结合蛋白。吸胀前体积的候选基因GRMZM2G318592和吸胀体积的候选基因GRMZM2G146173均编码包含锌指结构域的蛋白。吸胀前体积的候选基因GRMZM2G 449489和吸胀体积的候选基因GRMZM2G544352分别编码硒富集相关蛋白和未知功能蛋白。
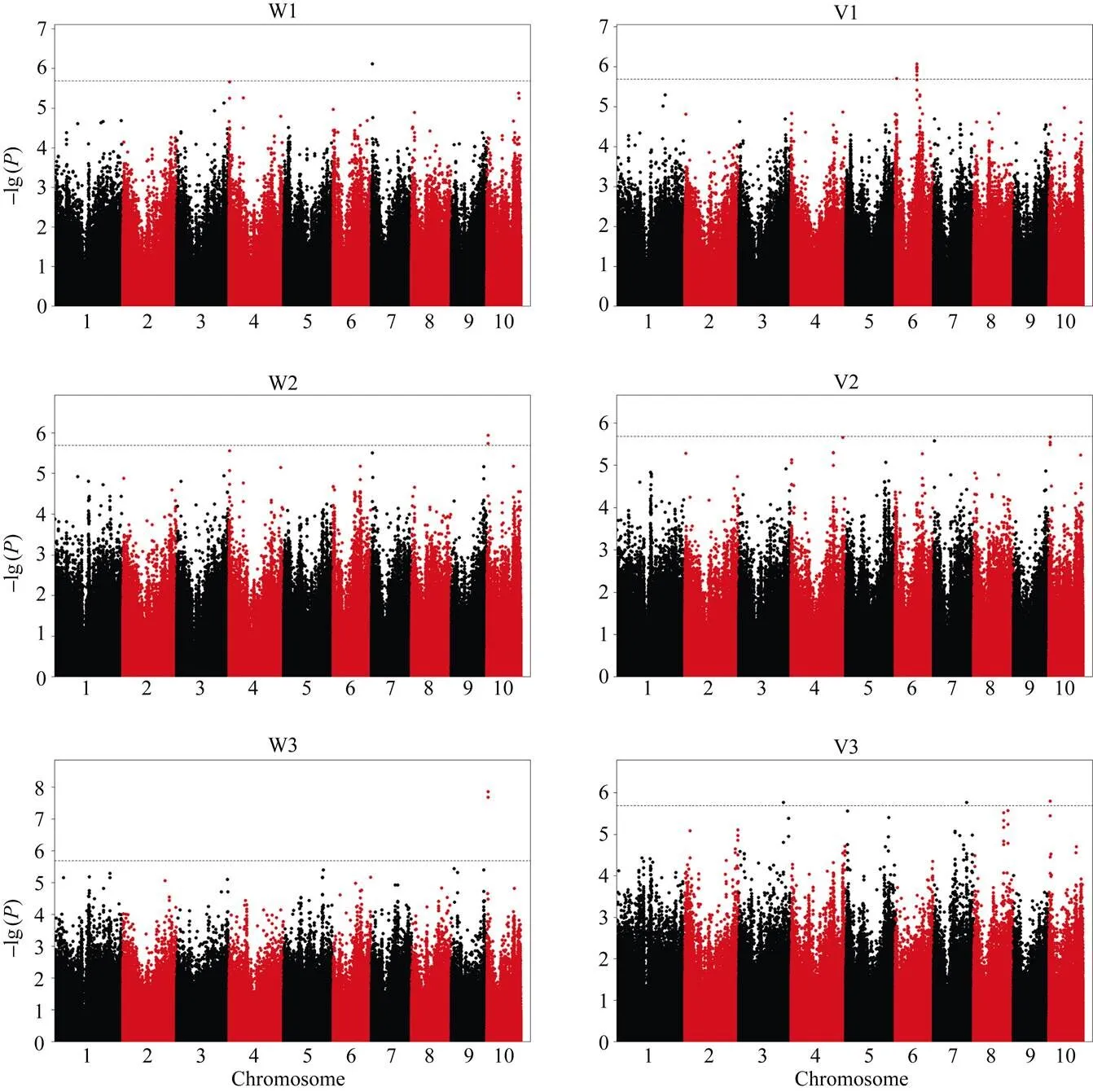
图3 种子萌发性状全基因组关联分析曼哈顿图
各性状详细名称见表1。黑色虚线代表全基因组关联分析的显著阈值。
Name of each trait is given in Table 1. Black dotted line indicates the genome-wide significance threshold.
3 讨论
不同的分析模型影响关联分析的结果。为确保关联结果的准确性, 最大程度降低假阳性结果, GWAS前要对每个性状进行最优模型的选择。Wang等[21]在玉米丝黑穗病的全基因组关联分析中发现, Q+K模型能更好地降低假阳性的发生。本研究对每个性状进行了3种模型(Q, K, Q+K)的分析, 发现Q+K模型对吸胀前重量、吸胀前体积、吸胀后重量、吸胀后体积和吸胀体积这5个性状的假阳性控制过于严格, 产生了一些假阴性的结果, K模型的拟合效果最好; 而Q+K模型则能更好的地控制吸胀重量性状的假阳性。
种子的萌发受赤霉素(GA)和脱落酸(ABA)等植物激素、光照、温度等环境因素以及一氧化氮(NO)和活性氧(ROS)等信号分子[22–27]在内的多种因素影响。本研究所筛选的候选基因的功能大多与种子内部激素水平、信号分子调控及活性氧有关。Li等[18]对水稻OSCA家族基因的功能研究证明钙离子可以通过调控吸胀种子的水势影响萌发, 因此与钙离子结合相关的结构或蛋白均可能影响种子萌发。GRMZM2G148411编码具有TLD结构域的核仁蛋白(TLD-domain containing nucleolar protein), 在拟南芥中有2个同源基因。其中, AT4G34070编码一种具有EF-hand基序的钙结合蛋白(Calcium-binding EF-hand family protein), 可能与NADPH氧化酶的合成有关; AT5G06260编码一种与影响钙离子结合的具有TLD结构域的核仁蛋白。GA可以作为主要的钙离子感受器, 增加钙离子和钙调蛋白, 对大麦糊粉层中钙离子信号传导起重要作用; ABA的作用与GA相反[28]。因此, 我们推测GRMZM2G148411可能作为NADPH氧化酶的组分, 调节种子内部ROS的浓度; 或作为钙离子调控的信号分子, 通过调节钙离子与钙调节蛋白的浓度影响种子内部GA和ABA的相对水平, 进而影响种子萌发。吸胀前体积候选基因GRMZM2G318592和吸胀体积候选基因GRMZM2G146173, 均编码一种含有锌指结构的蛋白。Baek等[29]发现, 拟南芥中的基因对ABA和氧化胁迫下的种子萌发和ROS含量具有重要作用, 该基因编码一种包含锌指结构域的核蛋白。Joseph等[30]研究拟南芥种子中ABA的合成与信号转导时, 发现ZFP3 (锌指蛋白3)是种子萌发过程中抑制ABA的调控因子, 赋予种子对ABA的不敏感性。因此, 推测本研究中的2个编码锌指蛋白的候选基因, 可能是通过调控玉米种子对ABA的敏感程度进一步影响种子的休眠和萌发。上述3个候选基因均作为信号分子直接或间接地参与种子萌发期间内部GA与ABA的相对水平以及ROS含量的调控, 进而影响种子的休眠与萌发。Yazdanpanah等[17]通过反向遗传学方法找到的种子休眠与萌发有关基因中, AT3G22490编码种子成熟蛋白(seed maturation protein), 该基因的敲除突变导致种子提前终止休眠。本研究中的候选基因GRMZM2G167892也编码一种种子成熟蛋白, 该基因可能与种子的萌发及休眠有关, 具体作用机制还有待进一步研究。
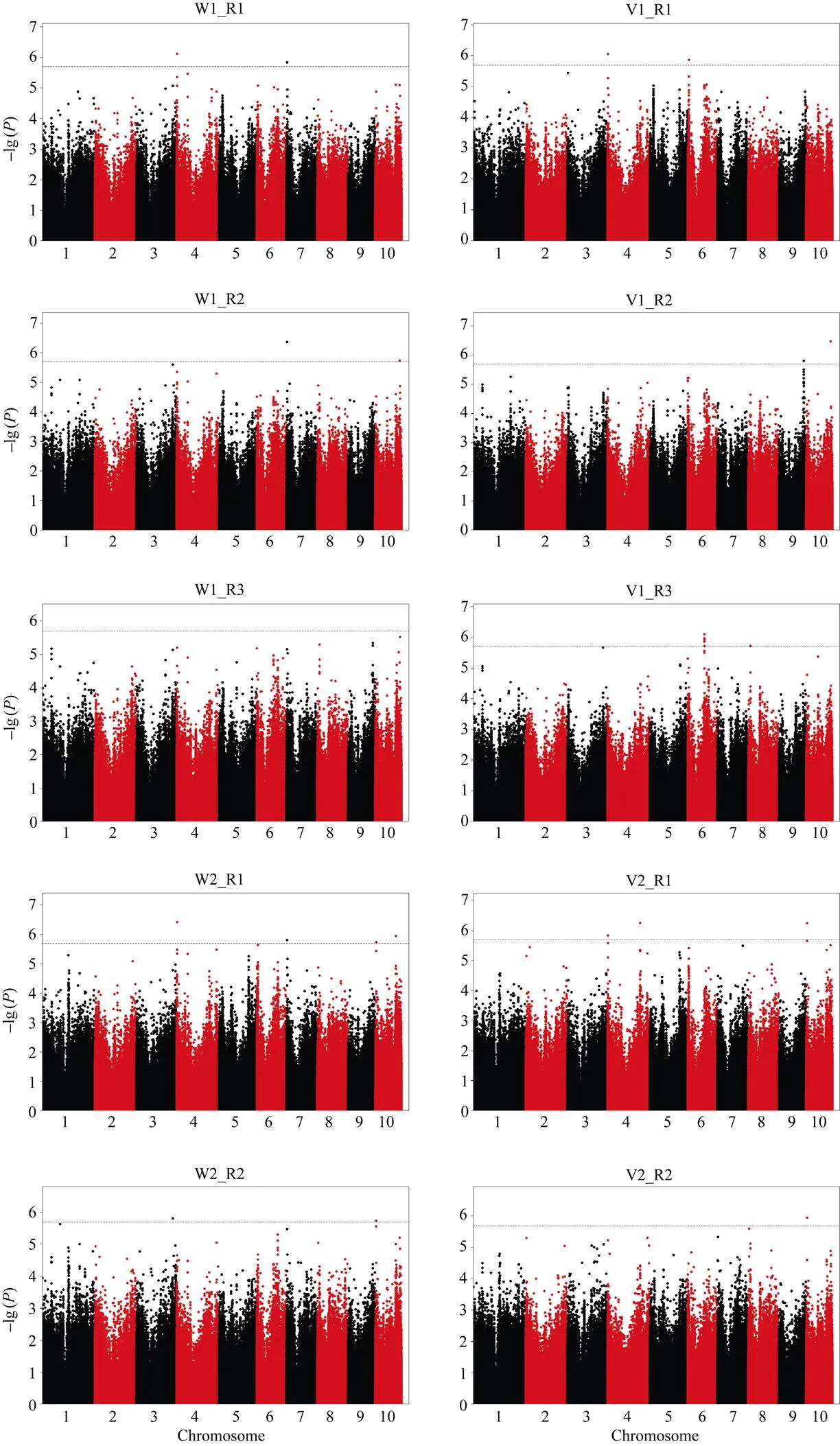
(图4)
Fig 4 Manhattan of GWAS for seed germination traits (single round)
各性状详细名称见表1。R1: 第1重复; R2: 第2重复, R3: 第3重复。黑色虚线代表全基因组关联分析的显著阈值。
Name of each trait is given in Table 1. R1: the first biological repeat; R2: the second biological repeat; R3: the third biological repeat. Black dotted line indicates the genome-wide significance threshold.
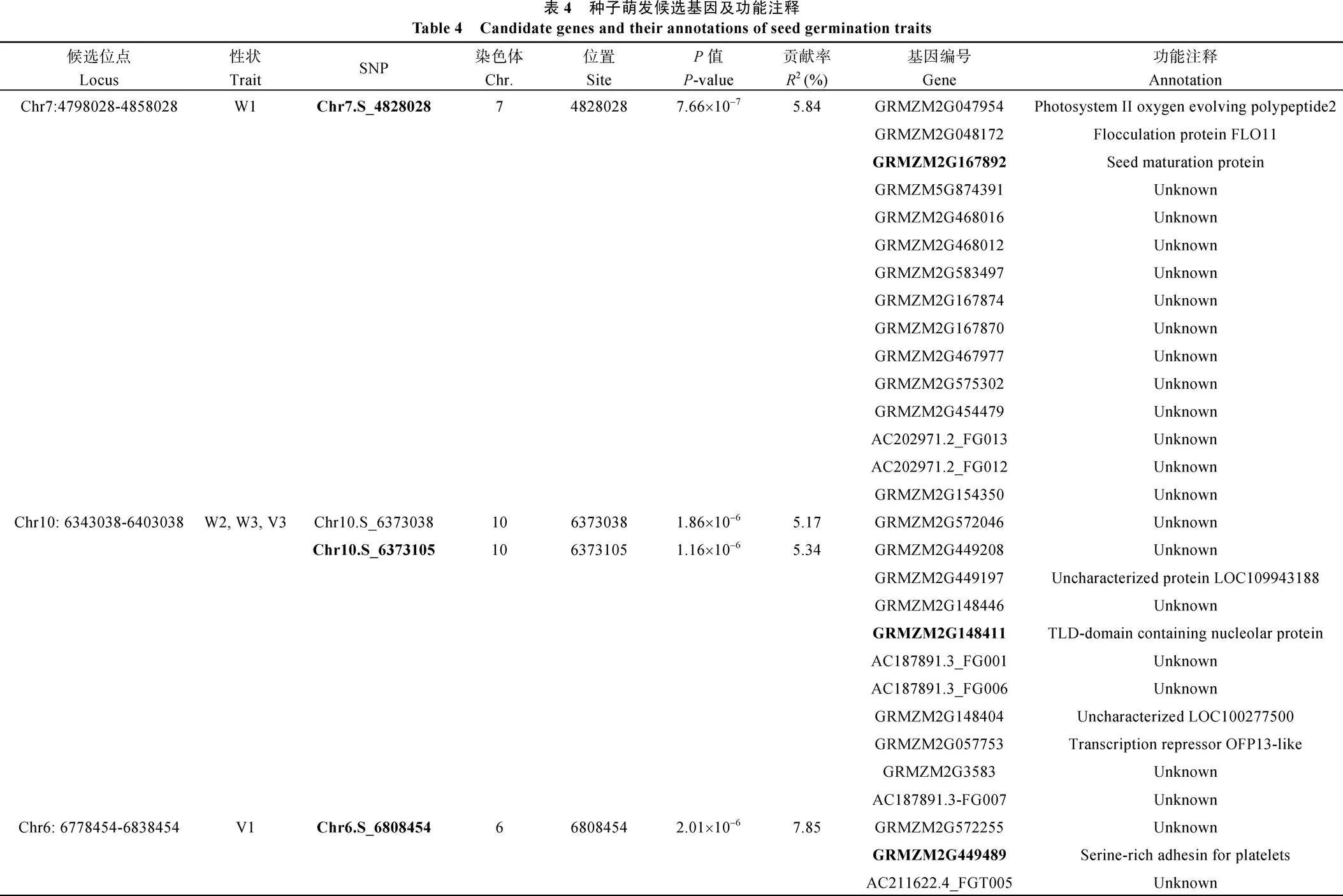
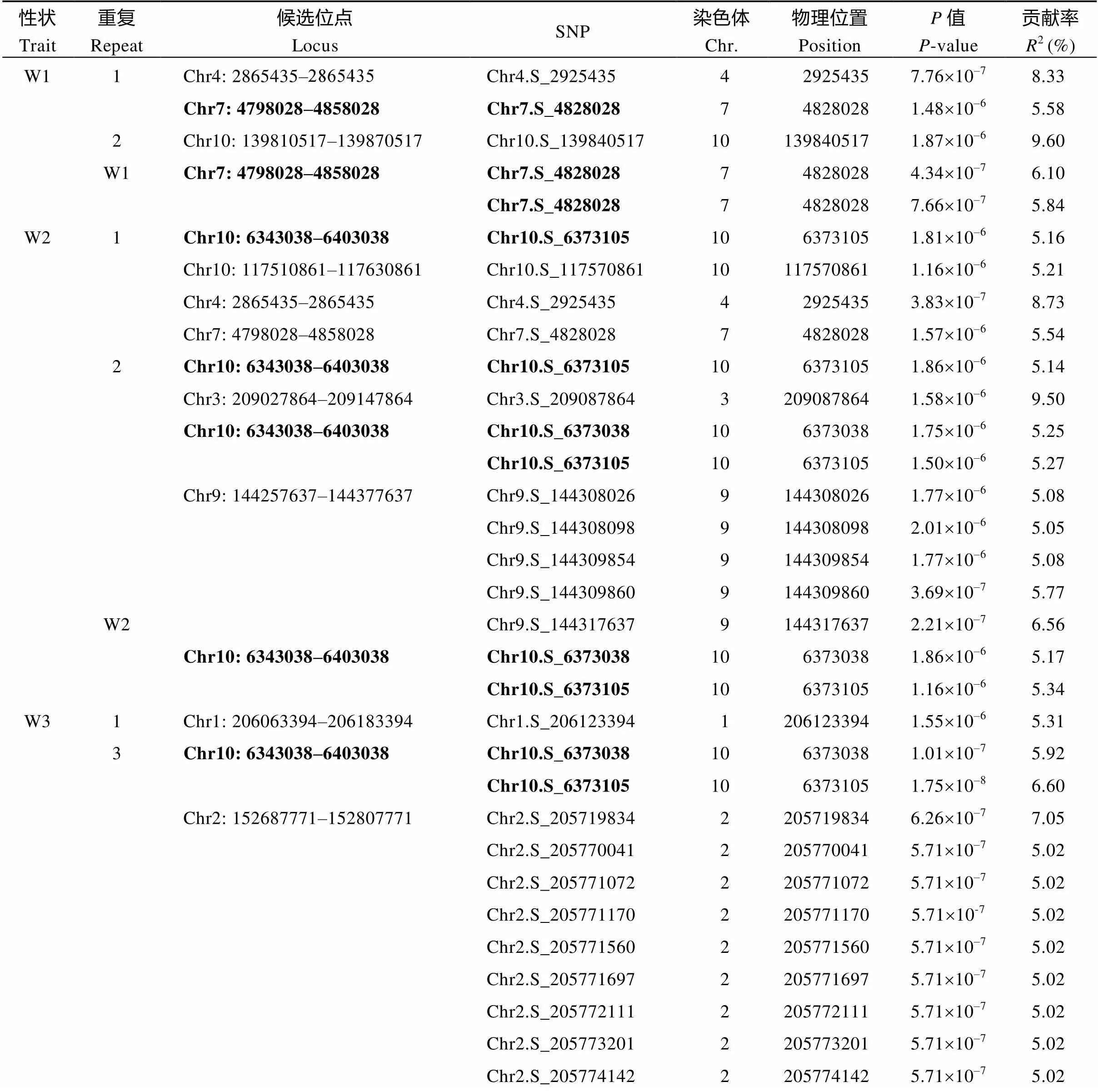
表5 种子萌发性状显著关联位点
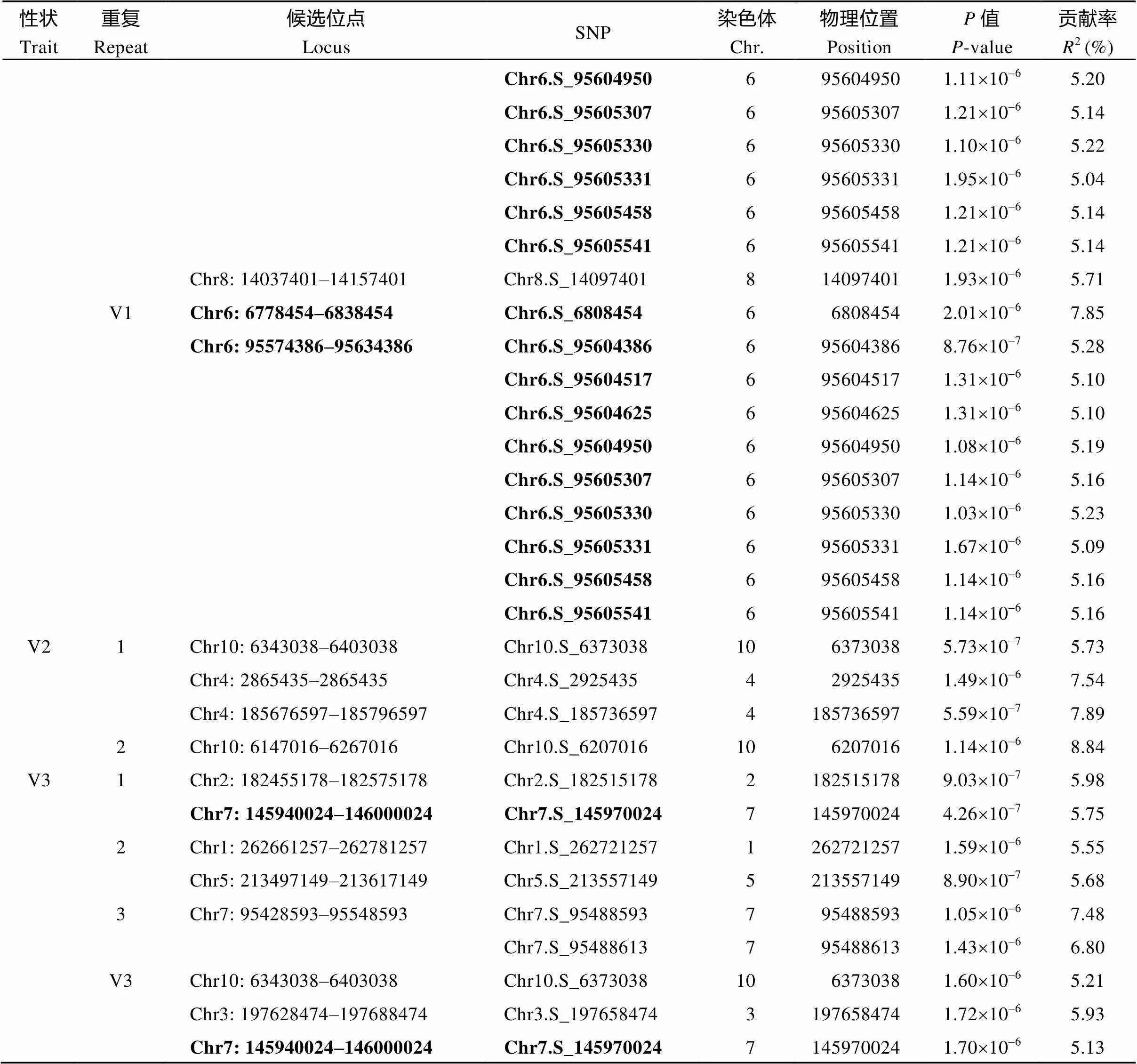
(续表5)
各性状详细名称见表1。每个性状中重复检测到的SNP用粗体标注。
Name of each trait is given in Table 1. SNPs repeatedly detected in each trait are marked boldly.
4 结论
共检测到15个显著的SNP-性状关联, 涉及6个QTL, 并鉴定出6个最可能的候选基因, 其中, 3个可能通过调节种子萌发过程中激素及ROS水平影响种子萌发, 1个可能通过调控种子成熟影响种子萌发, 另2个影响种子萌发的机制未知。
[1] Ellis R H. Seed and seedling vigor in relation to crop growth and yield., 1992, 11: 249–255
[2] Candela H, Hake S. The art and design of genetic screens: maize., 2008, 9: 192–203
[3] Below F E, Seebauer J R, Uribelarrea M, Schneerman M C, Moose S P. Physiological changes accompanying long-term selection for grain protein in maize. In: Janick J ed. Plant Breeding Reviews. New York: John Wiley & Sons, 2004. pp 133–151
[4] Klein R J, Zeiss C, Chew E Y, Tsai J Y, Sackler R S, Haynes C, Henning A K, SanGiovanni J P, Mane S M, Mayne S T, Bracken M B, Ferris F L, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration., 2005, 308: 385–389
[5] Atwell S, Huang Y S, Vilhjálmsson B J, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone A M, Hu T T, Jiang R, Muliyati N W, Zhang X, Amer M A, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker J R, Faure N, Kniskern J M, Jones J D, Michael T, Nemri A, Roux F, Salt D E, Tang C, Todesco M, Traw M B, Weigel D, Marjoram P, Borevitz J O, Bergelson J, Nordborg M. Genome-wide association study of 107 phenotypes in a common set ofinbred lines., 2010, 465: 627–631
[6] Huang X H, Zhao Y, Wei X H, Li C Y, Wang A H, Zhao Q, Li W J, Guo Y L, Deng L W, Zhu C R, Fan D L, Lu Y Q, Weng Q J, Liu K Y, Zhou T Y, Jing Y F, Si L Z, Dong G J, Huang T, Lu T T, Feng Q, Qian Q, Li J Y, Han B. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm., 2011, 44: 32–39
[7] Xiao Y J, Liu H J, Wu L J, Warburton M, Yan J B. Genome-wide association studies in maize: praise and stargaze., 2017, 10: 359–374
[8] Li H, Peng Z Y, Yang X H, Wang W D, Fu J J, Wang J H, Han Y J, Chai Y C, Guo T T, Yang N, Liu J, Warburton M L, Cheng Y B, Hao X M, Zhang P, Zhao J Y, Liu Y J, Wang G Y, Li J S, Yan J B. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels., 2013, 45: 43–50
[9] Liu H J, Luo X, Niu L Y, Xiao Y J, Chen L, Liu J, Wang X G, Jin M L, Li W Q, Zhang Q H, Yan J B. Distant eQTLs and non-coding sequences play critical roles in regulating gene expression and quantitative trait variation in maize., 2017, 10: 414-426
[10] Huang J, Zhang J H, Li W Z, Hu W, Duan L C, Feng Y Z, Qiu F Z, Yue B. Genome-wide association analysis of ten chilling tolerance indices at the germination and seedling stages in maize., 2013, 55: 735–744
[11] Shi Y Y, Gao L L, Wu Z C, Zhang X J, Wang M M, Zhang C S, Zhang F, Zhou Y L, Li Z K. Genome-wide association study of salt tolerance at the seed germination stage in rice., 2017, 17: 92
[12] Kan G Z, Zhang W, Yang W M, Ma D Y, Zhang D, Hao D R, Hu Z B, Yu D Y. Association mapping of soybean seed germination under salt stress., 2015, 290: 2147–2162
[13] Finch-Savage W E, Leubner-Metzger G. Seed dormancy and the control of germination., 2006, 171: 501–523
[14] Fu Z Y, Jin X N, Ding D, Li Y L, Fu Z J, Tang J H. Proteomic analysis of heterosis during maize seed germination., 2011, 11: 1462–1472
[15] Hu S D, Lübberstedt T, Zhao G W, Lee M. QTL mapping of low-temperature germination ability in the maize IBM Syn4 RIL population., 2016, 11: e0152795
[16] Han Z P, Ku L X, Zhang Z Z, Zhang J, Guo S L, Liu H V, Zhao R F, Ren Z Z, Zhang L K, Su H H, Dong L, Chen Y H. QTLs for seed vigor-related traits identified in maize seeds germinated under artificial aging conditions., 2014, 9: e92535
[17] Yazdanpanah F, Hanson J, Hilhorst H, Bentsink L. Differentially expressed genes during the imbibition of dormant and after- ripened seeds—a reverse genetics approach., 2017, 17: 151–162
[18] Li Y S, Yuan F, Wen Z H, Li Y H, Wang F, Zhu T, Zhuo W Q, Jin X, Wang Y D, Zhao H P, Pei Z M, Han S C. Genome-wide survey and expression analysis of thegene family in rice., 2015, 15: 261–273
[19] Noblet A, Leymarie J, Bailly C. Chilling temperature remodels phospholipidome ofseeds during imbibition., 2017, 7: 8886
[20] Deng M, Li D Q, Luo J Y, Xiao Y J, Liu H J, Pan Q C, Zhang X H, Jin M L, Zhao M C, Yan J B. The genetic architecture of amino acids dissection by association and linkage analysis in maize., 2017, 15: 1250–1263
[21] Wang M, Yan J B, Zhao J R, Song W, Zhang X B, Xiao Y N, Zheng Y L. Genome-wide association study (GWAS) of resistance to head smut in maize., 2012, 196: 125–131
[22] Batak I, Devic M, Giba Z, Grubisic D, Poff K L, Konjevic R. The effects of potassium nitrate and NO-donors on phytochrome A- and phytochrome B-specific induced germination ofseeds., 2002, 12: 253–259
[23] Bethke P C, Gubler F, Jacobsen J V, Jones R L. Dormancy of Arabidopsis seeds and barley grains can be broken by nitric oxide., 2004, 219: 847–855
[24] Bethke P C, Libourel I G, Reinohl V, Jones R L. Sodium nitroprusside, cyanide, nitrite and nitrate break Arabidopsis seed dormancy in a nitric oxide-dependent manner., 2006, 223: 805–812
[25] Sarath G, Bethke P C, Jones R, Baird L M, Hou G, Mitchell R B. Nitric oxide accelerates seed germination in warm-season grasses., 2006, 223: 1154–1164
[26] Fontaine O, Huault C, Pavis N, Billard J P. Dormancy breakage of Hordeum vulgare seeds: effects of hydrogen peroxide and scarification on glutathione level and glutathione reductase activity., 1994, 32: 677–683
[27] El-Maarouf-Bouteau H, Bailly C. Oxidative signaling in seed germination and dormancy., 2008, 3: 175–182
[28] Ishibashi Y, Kasa S, Sakamoto M, Aoki N, Kai K, Yuasa T, Hanada A, Yamaguchi S, Iwaya-Inoue M. A role for reactive oxygen species produced by NADPH oxidases in the embryo and aleurone cells in barley seed germination., 2015, 10: e0143173
[29] Baek D, Cha J Y, Kang S, Park B, Lee H J, Hong H, Chun H J, Kim D H, Kim M C, Lee S Y, Yun D J. The Arabidopsisa zinc finger domain protein ARS1 is essential for seed germination and ROS homeostasis in response to ABA and oxidative stress., 2015, 6: 963
[30] Joseph M P, Papdi C, Kozma-Bognár L, Nagy I, López-Carbonell M, Rigó G, Koncz C, Szabados L. The Arabidopsis ZINC FINGER PROTEIN3 interferes with abscisic acid and light signaling in seed germination and plant development., 2014, 165: 1203–1220
Genome-wide Association Studies of Seed Germination Related Traits in Maize
TIAN Run-Miao1,**, ZHANG Xue-Hai1,**, TANG Ji-Hua1, BAI Guang-Hong2, and FU Zhi-Yuan1,*
1College of Agronomy, Henan Agricultural University, Zhengzhou 450002, Henan, China;2Xinjiang Agricultural University, Urumqi 830052, Xinjiang, China
Germination is important for seed emergence, which has significant impact on maize yield. To reveal the genetic mechanism of maize seed germination, we investigated six traits related to seed germination using 467 diverse inbred lines. The genome-wide association studies (GWAS) between the six traits and 1.25M SNPs were implemented in three different models (Q model, K model, and Q+K model). The K model was much better than the other two models for weight before imbibition, volume before imbibition, weight after imbibition, volume after imbibition and volume of imbibition. While weight of imbibition trait could be well evaluated by Q+K model. In total, 15 SNPs were significantly associated with the six traits by the optimal model. These SNPs correspond to six QTLs, including five QTLs co-located in different biological replications. The six QTLs were located on chromosomes 3, 6, 7, and 10. The single SNP could explain 5.09%–7.85% variation of phenotype. Genes within or nearby most significant SNP were selected as candidates, and six candidate genes were identified for seed germination related traits in the six loci. Among these genes, GRMZM2G148411 encoding a TLD-domain calcium ion binding protein according to the annotations associated with weight after imbibition, volume after imbibition and volume of imbibition might be a signal molecule regulating seed germination and dormancy. The QTLs identified in this study are useful for developing functional markers and elucidating the genetic basis of seed germination.
maize; seed germination; genome-wide association study; candidate gene
2017-11-02;
2018-03-15;
2018-03-16.
10.3724/SP.J.1006.2018.00672
本研究由国家自然科学基金地区科学基金项目(31760389)资助。
This study was supported by the Regional Science Foundation of National Natural Science Foundation of China (31760389).
付志远, E-mail: fuzhiyuan2004@163.com
E-mail: tianrunmiao@foxmail.com
**同等贡献(Contributed equally to this work)
http://kns.cnki.net/kcms/detail/11.1809.S.20180316.0917.006.html
