固相萃取/定量核磁共振波谱法测定乳增宁胶囊中的淫羊藿苷
2018-05-10刘晓婷李苗苗郭强胜
刘晓婷,李苗苗,肖 坤,郭强胜,许 旭
(上海应用技术大学 化学与环境工程学院,上海 201418)
定量核磁共振波谱法(qNMR)相比于HPLC、GC-MS、LC-MS,具有无需待测物的高纯标准品等优势[1-2]。该法根据不同化学环境下质子NMR峰面积正比于质子数进行定量分析[3],是解决无标样定量分析问题的重要途径。近年来qNMR方法被广泛用于中药[4-7]、化学药[8-9]、食品[10]和代谢组学[11-13]等的研究。中国药典于2010版开始将该技术作为法定标准收载于通则(附录)中[14-15]。
乳增宁由艾叶、淫羊藿、天冬、柴胡、土贝母等多种中药组成[16],淫羊藿苷(Icariin)是处方中淫羊藿的主要活性成分[17-18]。近年来,对乳增宁胶囊中淫羊藿苷含量的测定已有不少研究,谢东等[19]将样品超声提取后采用柱层析和紫外法分析,汪涛等[20]、张玉婷等[21]超声提取后分别建立HPLC方法进行定量分析。但中药制剂成分复杂,样品中的强保留物对HPLC法柱子损伤较大[22]。李玮玲、李瑾翡等[23-24]分别使用固相萃取(SPE)后再用HPLC分析样品中的淫羊藿苷;干剑钦等[25]将肾宝合剂样品提取后用SPE柱处理后,用HPLC法分析其中的淫羊藿苷。这些HPLC方法在测定时均需要淫羊藿苷标样。
qNMR方法无需待测物的高纯标准品,只需常用的几种内标和氘代试剂即可完成定量分析[1],是常用中药分析方法的重要补充。但用于低含量成分分析时会存在灵敏度和干扰等问题。禹珊等[26]将两次丙酮提取后的样品水浴旋干后溶于少量氘代试剂,通过提高浓度来改善灵敏度。Yang等[7]使用氘代三氟乙酸使原本互相干扰的定量峰分离。
固相萃取(SPE)作为一种方便有效的样品前处理方法,具有净化样品和富集浓缩待测组分等功能。将SPE与qNMR结合,可以有效地解决qNMR的灵敏度和干扰问题。已有一些文献直接采用SPE/qNMR方法测定样品,如van der Hooft等[27]将尿液样品过HLB型SPE柱,4 mL甲醇洗脱后采用HPLC-TOF MS-SPE/NMR(1H NMR、2D-NMR)等对饮茶后人体尿液样品中的代谢物进行直接定性定量分析。Wagner等[28]使用SPE/qNMR分析污染工业废水中的原油和正己烷。Godejohann等[29]将SPE结合HPLC-UV与1H NMR对污染物进行了定量分析。但目前对SPE/qNMR联用方法进行系统研究、并做方法验证的文献报道不多。Pieri等[30]将样品提取物用C18型SPE柱净化富集,以dDMSO洗脱后用qNMR内标法测定了朝鲜蓟叶提取物中的莱蓟苦素。Moura[31]等将加入内标的样品溶液用C18型SPE进行富集净化后用qNMR测定了环境和生物样品中的β-N-甲基氨基-L-丙氨酸,并做了方法验证。Li等[32]将收集的42种党参样品提取后采用C18型SPE柱处理,用qNMR测定了4种生物碱。本课题组采用MCX型SPE柱结合qNMR定量测定了板蓝根饮片中的表告依春,并对重复性、精密度、线性、回收率等作了方法验证[33]。
本实验将SPE与qNMR结合,以乳增宁胶囊为样品,用SPE净化样品并富集浓缩待测组分含量,建立了SPE/qNMR测定乳增宁胶囊中有效成分淫羊藿苷含量的方法。对所建立方法的精密度、线性范围、灵敏度、回收率等进行了验证。本实验无需对照品即可实现对乳增宁胶囊中淫羊藿苷的定量测定。
1 实验部分
1.1 仪器与试剂
Bruker AVANCE Ⅲ 500MHz核磁共振谱仪(德国Bruker公司);U-3000高效液相色谱仪(Thermo-Fisher公司);M2p高精密天平(精度0.001 mg,德国Startoriu公司);AB204-N精密天平(精度0.1 mg,上海世义精密仪器有限公司);VGT-2120QT超声波清洗仪(广东固特超声实业有限公司);800B高速台式离心机(上海安亭科学仪器厂);DC-12氮吹仪(上海安谱科学仪器有限公司)。CNWBOND HC-C18固相萃取柱(2 g,10 mL,上海安谱实验科技股份有限公司);Innoval Neo XD高效液相色谱C18色谱柱(150 mm×4.6 mm,Agela Technologies)。
淫羊藿苷(Icariin,中国广州分析测试中心科力技术开发公司);2,3,5-三碘苯甲酸(>98%,上海阿拉丁生化科技股份有限公司);邻苯二甲酸氢钾(基准物质,99.95%~100.05%,上海泰坦科技股份有限公司);氘代二甲基亚砜(d6-DMSO,99.9%D,Cambridge Isotope Lab.Inc.);甲醇(HPLC级,阿达玛斯试剂有限公司);乙腈、乙醇为国产分析纯。实验室用水为屈臣氏蒸馏水。乳增宁胶囊样品1(蚌埠丰原涂山制药有限公司,批号:161104);样品2(修正药业集团长春高新制药有限公司,批号:160901);样品3(蚌埠丰原涂山制药有限公司,批号:161024)。
1.2 实验方法
1.2.1样品处理精密称取乳增宁胶囊内容物0.25 g,置于具塞锥形瓶中,加入10%乙醇10 mL,称重,超声提取40 min,取出放冷后再称重,加10%乙醇补充损失的质量。4 000 r/min离心5 min后,收集上层清液,过滤除去初滤液,收集续滤液。
依次用10 mL甲醇和10 mL水活化CNWBOND HC-C18SPE固相萃取柱(2 g,10 mL)。将6 mL上述续滤液过柱,加压控制流速大约为3秒/滴。之后用2 mL水对柱子进行淋洗。依次用2 mL乙腈和2 mL 45%乙腈进行洗脱。洗脱液用氮气吹至0.1 mL,加入0.4 mL氘代DMSO,再加入精密称取的2,3,5-三碘苯甲酸作为内标,混合均匀后转移至核磁管中。
1.2.2HPLC检测条件参考2015版中国药典[16]以及相关文献[25],选择用于考察SPE操作方法的HPLC检测条件为:Innoval Neo XD C18色谱柱(150 mm×4.6 mm),流动相甲醇-水(60∶40,体积比);流速1.0 mL/min;进样量10 μL;柱温35 ℃,检测波长270 nm。
1.2.3核磁共振定量核磁共振(NMR)采集条件:脉冲宽度P1=14.1 μs,延迟时间d1=1 s,扫描次数NS=256,谱宽SW=20 ppm,中心频率O1=3 090 Hz,采样时间AQ=3.17 s,于室温下测得核磁共振图谱。用Topspin软件将定量峰处的基线调平,然后选择相应质子峰的积分区间,将每个峰积分3~5次,当RSD < 1%时取平均值。采用2010版中国药典定量核磁共振技术的绝对定量公式(1)计算淫羊藿苷的含量[14-15]:
Ws=Wr×(As/Ar)×(Es/Er)
(1)
式中,Wr为内标物的质量,Es为供试品的质子当量重量(分子量除以特征峰的质子数),Er为内标物的质子当量重量,As为供试品的峰面积,Ar为内标峰的峰面积。
2 结果与讨论
2.1 样品制备方法的选择
2.1.1样品提取条件的优化乳增宁中淫羊藿苷的提取方法以超声波提取为主[19-21],本实验用“1.2.2”的HPLC检测条件考察了提取次数影响。称取乳增宁胶囊内容物0.25 g,按“1.2.1”对样品进行前处理后,检测续滤液。结果显示,第2次超声后续滤液中淫羊藿苷的峰面积仅为第1次提取的1/70。因此本实验选择1次超声提取。
中国药典中对淫羊藿苷采用超声提取(功率360 W,频率50 kHz) 40 min[16]。本实验超声仪器功率为400 W,根据药典分别考察超声提取10、20、30、40、50、60 min对淫羊藿苷提取效果的影响。根据峰面积估计淫羊藿苷的含量。结果显示,超声40 min时,直接HPLC测定的淫羊藿苷峰面积较大,继续增加时间响应值(峰面积)差异不大。故选择超声时间为40 min。
综合以上实验,最终选择乳增宁胶囊中淫羊藿苷的提取方法为:40 min超声提取1次。弃去前2 mL初滤液,取续滤液6 mL过固相萃取小柱。
2.1.2固相萃取条件的选择优化了SPE柱的类型及其活化、样品溶剂类型、上样量、柱淋洗剂的类型、柱洗脱剂的类型、洗脱剂用量7个条件,以选择合适的固相萃取条件。
SPE柱及其活化:文献中[23-25]多采用C18柱纯化淫羊藿苷,本实验使用C18柱也取得了较好的效果,因此选择C18柱。通过比较,选择用10 mL甲醇与10 mL水活化平衡C18固相萃取柱。
样品溶剂的选择:考察了30%乙腈、60%甲醇、水、5%乙醇、10%乙醇、70%乙醇6种溶剂溶解乳增宁胶囊内容物时对淫羊藿苷的保留效果。结果显示,用30%乙腈、60%甲醇、70%乙醇作为样品溶剂进行上样时均有大量的淫羊藿苷漏下。用水、5%乙醇与10%乙醇作为溶剂上样时对淫羊藿苷的保留效果最好,但由于水与5%乙醇对于乳增宁中淫羊藿苷的提取率比10%乙醇差,所以实验选用10%乙醇。
上样量:将8 mL续滤液过柱,分别接第2、4、6、8 mL流出液进行检测,前6 mL淫羊藿苷均能够很好地保留在柱上,后2 mL会有少量淫羊藿苷漏出,所以最终选择上样量为6 mL。
淋洗剂:选择2 mL水进行淋洗,并将流出液收集进行检测,均未检测到淫羊藿苷。
洗脱剂:考察了用甲醇、乙腈、45%乙腈洗脱淫羊藿苷和后续吹干的效果,发现用相同体积的甲醇或乙腈不能完全洗脱淫羊藿苷,最终选择用乙腈与45%的乙腈相结合的方法进行洗脱。
洗脱剂用量:先用乙腈2 mL洗脱,然后用45%乙腈4 mL分2次,每次2 mL继续洗脱,收集洗脱液,并对洗脱液进行检测。结果显示,第5~6 mL洗脱液中淫羊藿苷的峰面积大约为第1~2 mL洗脱液的0.3%,因此本实验选择的洗脱剂用量为乙腈和45%乙腈各2 mL。
综上所述,固相萃取的最终条件为:活化:10 mL甲醇、10 mL水;上样:6 mL 10%乙醇;淋洗:2 mL水;洗脱:2 mL乙腈,2 mL 45%乙腈。
2.2 氘代试剂的选择
将得到的4 mL洗脱液氮气吹干后用不同的氘代溶剂溶解,氘代DMSO能很好地溶解产物,并对定量峰无干扰,因此实验选择氘代DMSO作为溶剂。
2.3 内标物的选择
考察了几种常用的内标物(邻苯二甲酸氢钾、苯甲酸和2,3,5-三碘苯甲酸)的效果,其中前两者的NMR峰均会与样品NMR峰有重叠,而2,3,5-三碘苯甲酸与样品组分互相不干扰,因此选择2,3,5-三碘苯甲酸作为内标物。
2.4 信号归属及定量峰的确认
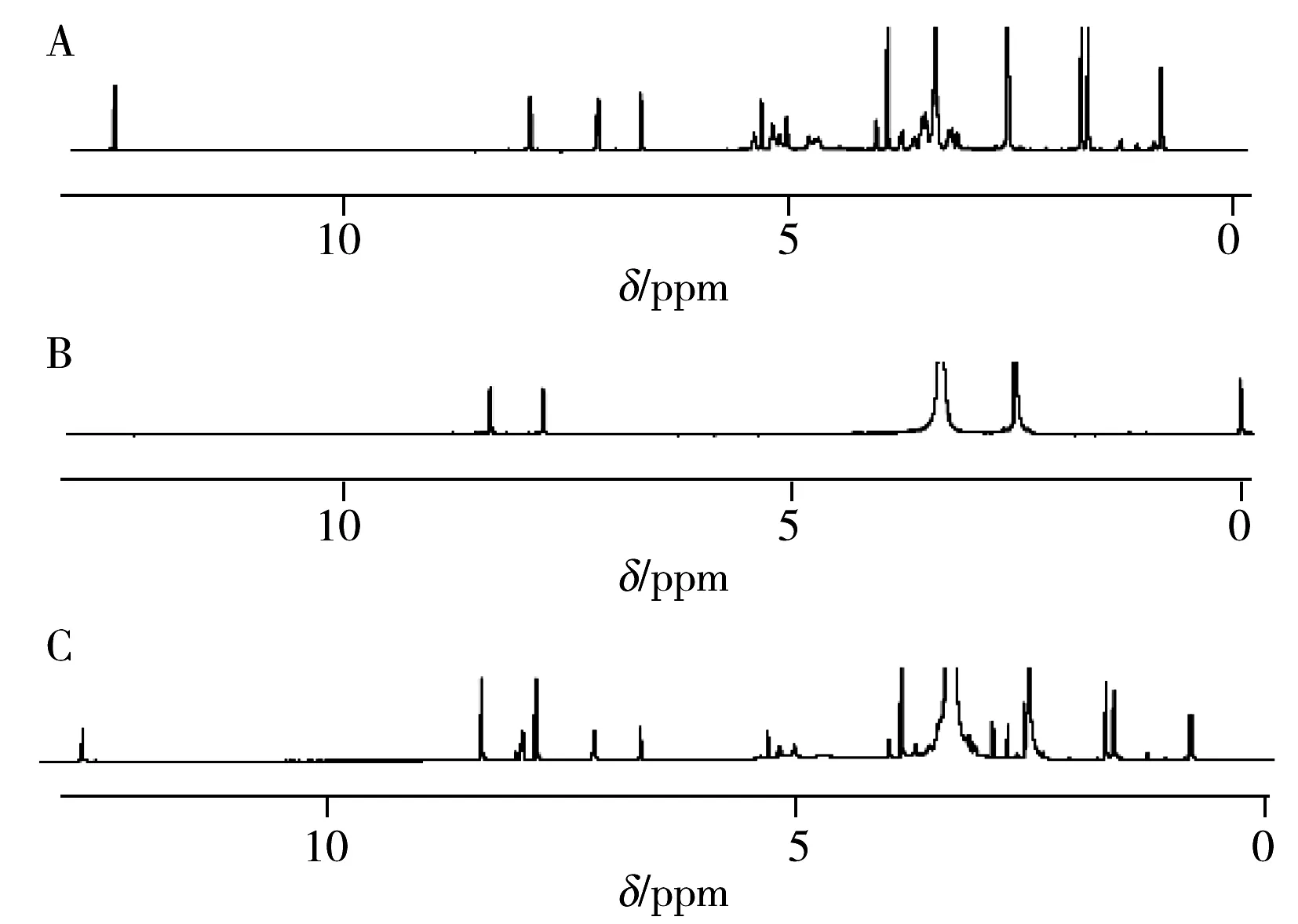
图1 淫羊藿苷标液与内标2,3,5-三碘苯甲酸的1H NMR谱图Fig.1 NMR spectra of icariin standard and 2,3, 5-triiodobenzoate internal standardA.icariin;B.2,3,5-triiodobenzoate;C.the mixture of A and B
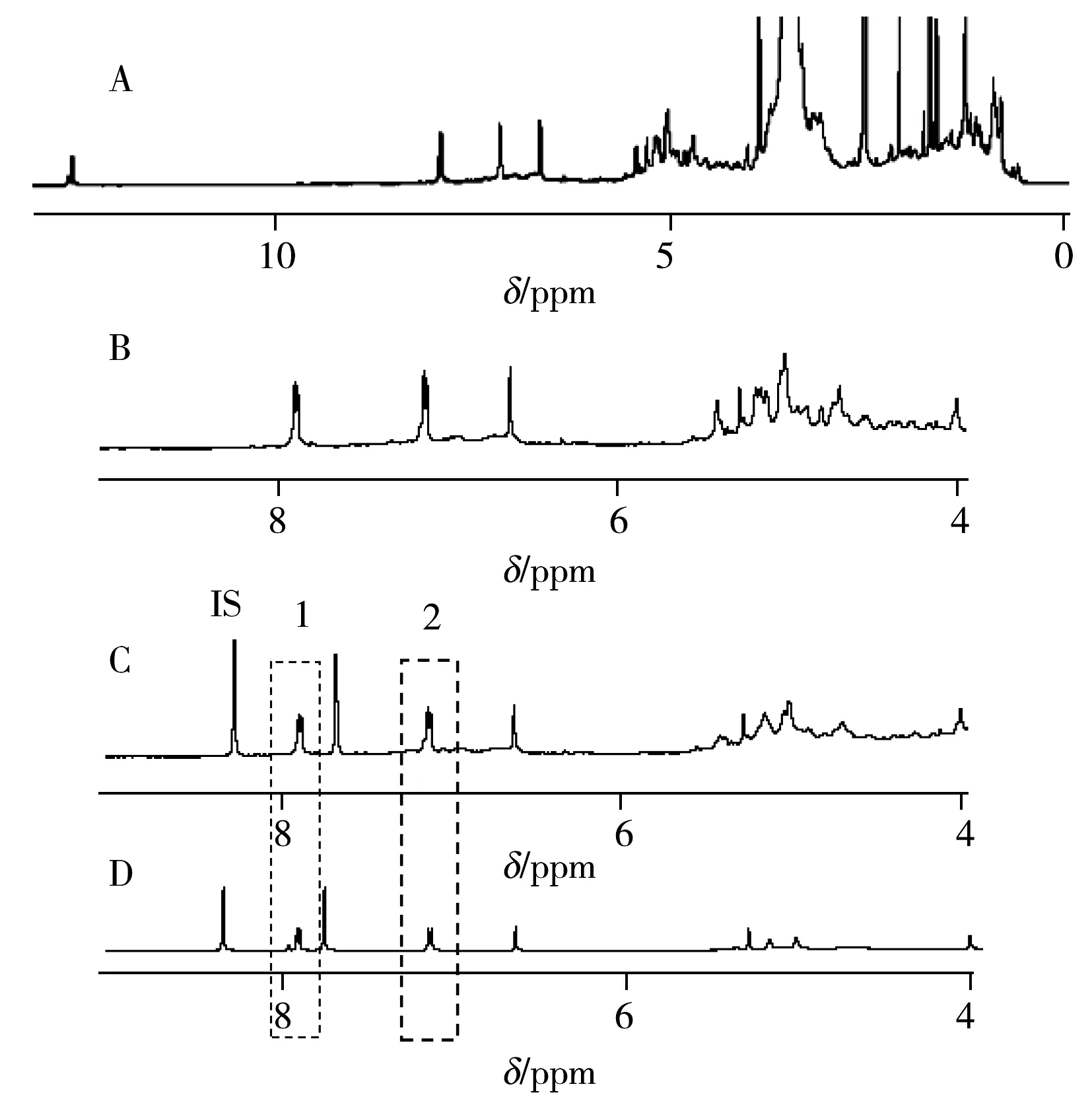
图2 乳增宁胶囊样品及其加标混合物的1H NMR谱图Fig.2 NMR spectra of Ruzengning capsule sample and mixed chemical referencesA.sample;B.the enlarge part of Fig.A;C.mixture of sample and 2,3,5-triiodobenzoate;D.mixture of icariin and 2,3,5-triiodobenzoate
2.4.1NMR峰归属采用“1.2.3”方法分别测定淫羊藿苷标准品、内标2,3,5-三碘苯甲酸以及两者混合物的NMR谱图(见图1)。对比图1中的核磁图,并参考文献[34],归属混合物(图1C)中的各信号峰。
淫羊藿苷的峰:δ1.607(5”-H,s,3H);δ1.691(4”-H,s,3H);δ3.861(OCH3,s,3H);δ5.013(Glc-1-H,d,1H);δ5.165(2”-H,t,1H);δ5.287(Rha-1-H,d,1H);δ6.638(6-H,s,1H);δ7.126~7.143(3’,5’-H,d,2H);δ7.890~7.909(2’,6’-H,d,2H);δ12.574(5-OH,s,1H)。
2,3,5-三碘苯甲酸峰:δ7.749~7.751(d,1H);δ8.330~8.338(d,1H)。
2.4.2定量峰的确认采用“1.2”方法,测得样品1的NMR图谱见图2A,样品和内标物混合物的NMR谱图见图2D。
qNMR定量峰需满足分离度较好,信噪比≥150等要求。图2C中识别的两个淫羊藿苷的特征峰1和2均能满足要求,由于峰1处的基线较平,更适合做定量峰,因此选择δ7.890~7.909(2’,6’-H,d,2H)(图2C峰1)作为定量峰。同样,根据条件选择δ8.330~8.338(d,1H)作为内标的定量峰(图2C峰IS)。
2.4.3核磁实验参数的影响分别考察了脉冲宽度P1、延迟时间d1和扫描次数NS对积分面积的影响。当P1≥14.1 μs,d1=1 s,NS≥256时,淫羊藿苷定量峰和内标定量峰的信噪比可以达到定量要求(S/N≥150),且测定的Ar与As值趋于稳定。故实验选定核磁参数P1=14.1 μs,d1=1 s,NS=256。
2.4.4对2,3,5-三碘苯甲酸及淫羊藿苷纯品的含量校正实验使用的淫羊藿苷和内标采用qNMR法进行含量校正。在“1.2.3”实验条件下,以DMSO为溶剂,基准物质邻苯二甲酸氢钾的NMR峰H-4,5(7.502~7.520,dd,2H)与三碘苯甲酸的NMR定量峰互不干扰。所以选择邻苯二甲酸氢钾作为校准2,3,5-三碘苯甲酸(定量峰为H-4(δ7.695~7.699,d,1H ))和淫羊藿苷(定量峰δ7.126~7.143(3’,5’-H,d,2H))含量的内标物质。采用定量核磁共振技术的绝对定量模式对内标和淫羊藿苷纯品进行含量校正。重复试验6次,取平均值。实验测得内标的含量为99.3%,RSD为0.59%;淫羊藿苷纯品含量为96.4%,RSD为0.65%。以下实验使用校正后的含量值进行。
2.5 方法验证
2.5.1精密度取样品1按照“1.2.1”方法制成待测样品,分别在日内和日间各做6组测试,以淫羊藿苷定量峰δ7.890~7.909(2’,6’-H,d,2H)与内标峰δ8.330~8.338(d,1H)的积分面积(As/Ar)为指标,验证qNMR方法的精密度。日内RSD为0.43%,日间RSD为0.75%。表明定量核磁共振技术的精密度好,样品的稳定性好。
2.5.2线性与分析性能称取6组淫羊藿苷(0.403 3~2.111 mg)和1.5 mg内标,溶解于0.5 mL氘代DMSO中,超声溶解后,按“1.2”方法获得核磁共振谱图。选择淫羊藿苷与2,3,5-三碘苯甲酸质量比(x)为横坐标,核磁共振图谱中淫羊藿苷与2,3,5-三碘苯甲酸峰面积比(y)为纵坐标,进行线性拟合。得到标准曲线为y=1.432 7x,相关系数r=0.999 9。
根据“1.2.3”公式(1)理论计算出淫羊藿苷线性曲线的斜率(Es/Er)为1.477 3,与实验中测得淫羊藿苷标准曲线的斜率值接近,说明在实际样品测定时,可以采用qNMR技术的绝对定量公式来直接计算淫羊藿苷的含量,而不必使用对照品。
2.5.3检出限与定量下限根据2015版中国药典四部通则[14],qNMR的检出限(LOD)和定量下限(LOQ)由基于响应值标准偏差和标准曲线斜率的公式LOD = 10δ/S,LOQ = 3.3δ/S得到,式中δ采用标准曲线的剩余标准偏差来计算。得淫羊藿苷的LOD=0.020 3 mg,LOQ=0.061 4 mg。本实验中内标的用量约为1.5 mg,溶剂0.5 mL计算得淫羊藿苷的LOD=0.061 g/L,LOQ=0.184 g/L。英国药典[35]对qNMR方法要求信噪比S/N≥150∶1,这也被认为是LOQ的指标,以样品1的NMR测定谱峰信噪比,根据定量核磁共振技术的绝对定量公式[14-15]计算得淫羊藿苷的LOQ为0.173 g/L。综上,可以认为本方法对淫羊藿苷的LOQ为0.184 g/L,LOD为0.061 g/L。按照本实验中约0.5 mL溶剂的量和约0.25 g乳增宁的样品量,可算出本法测定乳增宁胶囊中淫羊藿苷含量的LOD为0.122 mg/g;LOQ为0.368 mg/g,相当于0.036 8%(质量分数)。
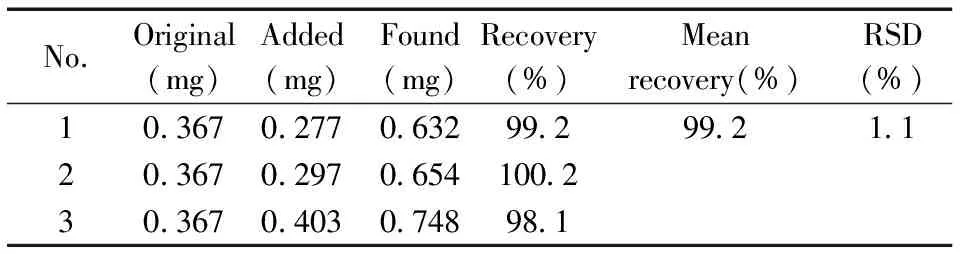
表1 样品提取后的qNMR方法回收率(n=3)Table 1 Recovery of icariin by qNMR after the sample extraction(n=3)
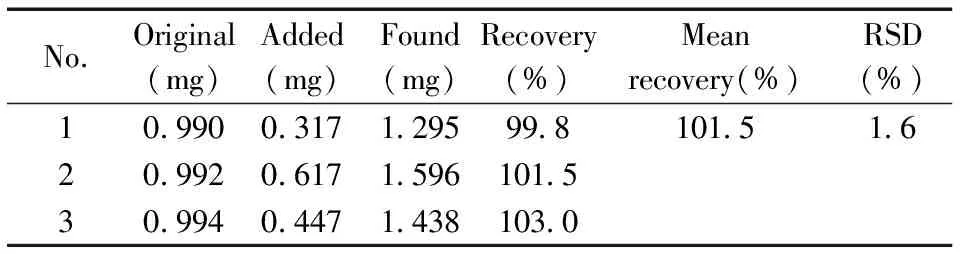
表2 包括样品提取过程的SPE/qNMR方法回收率(n=3)Table 2 Recovery of icariin including the extraction process(n=3)
2.5.4样品提取后qNMR方法的回收率取样品用“1.2”方法测定其中淫羊藿苷的含量,然后将相同的样品提取溶液再取3份,在每份溶液中加入淫羊藿苷标准品(淫羊藿苷对照品含量为96.4%)后再测定含量,计算除去样品提取过程的qNMR方法的加标回收率,结果见表1。淫羊藿苷的回收率为98.1%~100.2%,RSD为1.1%。
2.5.5包括样品提取过程的SPE/qNMR方法回收率分别准确称取3份0.25 g样品1,每份均加入一定量的淫羊藿苷标准品,用“1.2”提取分析方法对样品进行定量。结果如表2,得到包括样品SPE预处理过程的淫羊藿苷的回收率为99.8%~103.0%,RSD为1.6%。
2.6 样品分析
按“1.2”方法,称取不同厂家和批号不同的乳增宁胶囊内容物,重复测定3次样品1、2、3的平均值分别为3.95、4.39、4.11 mg/g,RSD分别为1.9%、1.4%和0.75%。按照2015版中国药典每粒乳增宁胶囊中的淫羊藿苷的测定不低于1.5 mg/g[16]的规定,3个样品均符合要求。
3 结 论
建立了SPE/qNMR测定乳增宁胶囊中淫羊藿苷含量的方法,将样品通过SPE进行富集纯化,改善了qNMR的检测灵敏度。方法精密度好,定量分析结果可靠,在实际样品测定时,可以不用淫羊藿苷对照品,直接以qNMR绝对定量公式来计算淫羊藿苷的含量。
参考文献:
[1] Pauli G F,Gödecke T,Jaki B U,Lankin D C.J.Nat.Prod.,2012,75(4):834-851.
[2] Simmler C,Napolitano J G,McAlpine J B,Chen S N,Pauli G F.Curr.Opin.Biotechnol.,2014,25:51-59.
[3] Liu Y,Hu C Q.Chin.J.Pharm.Anal.(刘英,胡昌勤.药物分析杂志),2001,21(6):447-452.
[4] Zou P P,Tu P F,Jiang Y.Anal.Methods,2013,5(4):1062-1067.
[5] Liu N Q,Choi Y H,Verpoorte R,Kooy F.Phytochem.Anal.,2010,21(5):451-456.
[6] Fan G,Zhang M Y,Zhou X D,Lai X R,Yue Q H,Tang C,Luo W Z,Zhang Y.Anal.Chim.Acta,2012,747:76-83.
[7] Yang M,Wang J,Kong L J.Pharm.Biomed.Anal.,2012,70:87-93.
[8] Parmar K,Mahato A,Patel R,Prajapati S.Int.J.ChemTechRes.,2013,5(1):312-321.
[9] Gadape H,Parikh K.Anal.Methods,2011,3(10):2341-2347.
[10] López-Rituerto E,Cabredo S,López M,Avenoza A,Busto J H,Peregrina J M.J.Agric.FoodChem.,2009,57:2112-2118.
[11] Shen G P,Dai X X,Deng L L,Wei Y Y,Xu J J,Wang C H,Dong J Y,Chen Z.Chem.J.Chin.Univ.(沈桂平,戴晓侠,邓伶莉,危阳洋,许晶晶,王彩虹,董继扬,陈忠.高等学校化学学报),2013,10(34):2290-2295.
[12] Barding Jr G A,Salditos R,Larive C K.Anal.Bioanal.Chem.,2012,404(4):1165-1179.
[13] Salem A A,Abdou I M,Saleh H A.J.AOACInt.,2012,95(6):1644-1651.
[14] National Pharmacopoeia Committee.ThePharmacopoeiaofthePeople′sRepublicofChina(PartⅣ,2015 Ed).Beijing:China Medical Science Press(国家药典委员会.中华人民共和国药典(四部,2015年版).北京:中国医药科技出版社),2015:52-54,376.
[15] National Pharmacopoeia Committee.ThePharmacopoeiaofthePeople′sRepublicofChina(PartⅡ,2010 Ed).Beijing:China Medical Science Press(国家药典委员会.中华人民共和国药典(二部,2010年版).北京:中国医药科技出版社),2010:Appendix Page 81-83.
[16] National Pharmacopoeia Committee.ThePharmacopoeiaofthePeople′sRepublicofChina(PartⅠ,2015 Ed).Beijing:China Medical Science Press(国家药典委员会.中华人民共和国药典(一部,2015年版).北京:中国医药科技出版社),2015:327-328,1098.
[17] Feng X Y,Yan L P,Yang Y.LishizhenMed.Mater.Med.Res.(冯晓燕,严鲁萍,杨源.时珍国医国药),2007,18(12):3070-3072.
[18] Jia L L,Yuan D,Wang H W,He Y M,Zhang C C.Prog.Mod.Biomed.(贾亮亮,袁丁,王洪武,何毓敏,张长城.现代生物医学进展),2010,10(20):3976-3979.
[19] Xie D,Yang J J.TianjinPharm.(谢东,杨戒骄.天津药学),2001,13(4):84-85.
[20] Wang T,Zhao Y P,Hu W D.Chin.J.DrugAppl.Monitor.(汪涛,赵雁平,胡卫东.中国药物应用与监测),2016,3(5):271-273.
[21] Zhang Y T,Gao J R,Liu J,Jiang H,Wu J.Clin.J.Tradit.Chin.Med.(张玉婷,高家荣,刘健,姜辉,吴健.中医药临床杂志),2016,(4):559-561.
[22] Yang S,Jin B F,Xu M.Chin.J.Pharm.Anal.(杨松,靳宝峰,许敏.药物分析杂志),1995,15(Suppl):105.
[23] Li W L.GuangdongPharm.J.(李玮玲.广东药学),2005,15(1):1-3.
[24] Li J F,Fang J H.Tradit.Chin.DrugRes.Clin.Pharmacol.(李瑾翡,方继辉.中药新药与临床药理),2001,(4):287-289.
[25] Gan J Q,Yin X Y.J.JiangxiUniv.Tradit.Chin.Med.(干剑钦,尹小英.江西中医学院学报),2004,(50):49-50.
[26] Yu S,Guo Q S,Wang H L,Gao J P,Xu X.Chin.J.Anal.Chem.(禹珊,郭强胜,王会琳,高建平,许旭.分析化学),2015,43(1):69-74.
[27] van der Hooft J J J,de Vos R C H,Mihaleva V,Bino R J,Ridder L,de Roo N,Jacobs D M,van Duynhoven J P M,Vervoort J.Anal.Chem.,2012,84(16):7263-7271.
[28] Wagner L,Kall C,Fridjonsson E O,May E F,Stanwix P L,Graham B F,Carroll M R J,Johns M L.Meas.Sci.Technol.,2016,27(10):105501.
[29] Godejohann M,Heintz L,Daolio C,Berset J D,Muff D.Environ.Sci.Technol.,2009,43(18):7055-7061.
[30] Pieri V,Stuppner H.Plant.Med.,2011,77(15):1756-1758.
[31] Moura S,Ultramari M D A,Paula D M L D,Yonamine M,Pinto E.Toxicon,2009,53(5):578-583.
[32] Li C Y,Xu H X,Han Q B,Wu T S.J.Chromatogr.A,2009,1216(11):2124-2129.
[33] Liu X T,Yu S,Yuan M,Guo Q S,Gong C,Xu X.Chin.J.Anal.Chem.(刘晓婷,禹珊,袁铭,郭强胜,龚灿,许旭.分析化学),2017,45(7):1059-1065.
[34] Li F,Liu Y L.ActaPharm.Sin.(李枫,刘永隆.药学学报),1998,23(10):739-748.
[35] The British Pharmacopoeia Commission.BritishPharmacopoeia(2013 Ed).London:The Stationary Office,2013:Appendix IIC Nuclear Magnetic Resonance Spectrometry.
