c-Jun氨基末端激酶通路在缺氧性肾小管上皮细胞转分化中的作用
2018-05-10江星伯蒋玲龚玲宋坤岭余雪云覃远汉
江星伯 蒋玲 龚玲 宋坤岭 余雪云 覃远汉
广西医科大学第一附属医院儿科(南宁 530021)
肾间质纤维化(renal interstitial fibrosis,RIF)的关键因素和中心环节是肾小管上皮细胞(renal tubular epithelial cell,RTEC)损伤和功能不全[1],缺氧是引起RIF发生发展的一个重要微环境因素,丝裂原活化蛋白激酶(mitogen-activiated protein kinases,MAPK)超家族中的应激活化蛋白激酶(C-jun N-terminal kinase,JNK)可在缺氧条件下被激活[2]。有研究表明[3-5],在肾小管损伤中存在大量JNK的激活,JNK可能在缺血缺氧损伤中发挥重要作用。α-平滑肌肌动蛋白(α-smooth muscle actin,α-SMA)是肌成纤维细胞(myofibroblast,MF)的标志性蛋白,RIF常出现肾脏固有细胞表型转分化,大量表达α-SMA,因此α-SMA的表达变化在RIF中具有重要意义。HONMA等[6]报道,肾脏组织的JNK蛋白的表达量与组织纤维化和肾功能不全呈正相关。JNK在体外培养的大鼠RTEC缺氧性损伤中对α-SMA的作用,国内尚未发现相关研究报道,为此我们通过检测JNK、pJNK和α-SMA在大鼠RTEC缺氧性损伤表达变化,初步探讨JNK通路在缺氧性RTEC转分化中的作用,为防治RIF提供新思路。
1 材料与方法
1.1 实验材料大鼠肾小管上皮细胞株(NRK-52E)购自上海生物细胞库。DMEM basic(1×)培养基、混合胎牛血清均购自美国Gibco公司,青、链霉素和JNK通路特异性阻断剂SP600125均购自碧云天公司,缺氧真空罐购自加拿大Stemcell公司,Trizol RNA提取试剂盒购自美国Invitrogen公司,RevertAid First Strand cDNA Synthesis Kit试剂盒购自美国Thermo公司,FastStart Universal SYBR Green Master(Rox)试剂盒购自德国 Roche公司,5×电泳液、10×电转液、20×TBS溶液、Tween-20均购自北京索莱宝公司,PVDF膜购自美国Immobilon-P公司,BCA蛋白浓度测定试剂盒购自南京建成生物工程研究所,引物设计及合成均由宝生物工程(大连)有限公司完成,JNK抗体、pJNK抗体和β-actin抗体购自美国CST公司,α-SMA抗体购自美国Abcam公司,辣根过氧化物酶标记的二抗购自美国CST公司,Chemiluminescent HRP Substrate购自美国Millipore公司,FluorChem M购自美国Protein Simple公司,实时荧光定量PCR仪7500购自美国Applied Biosystems公司。
1.2 上皮细胞培养与缺氧损伤模型构建大鼠肾小管上皮细胞(NRK-52E)培养在7%FBS、100 U/mL青霉素和100 μg/mL链霉素的DMEM培养基中,在37℃、50 mL/L二氧化碳(CO2)培养箱中培养,细胞生长至约70%融合时传代。待传代的细胞生长至对数期时,将细胞随机分为5组:正常组、缺氧组、抑制剂组、激动剂组、DMSO组。正常组在正常培养条件下继续培养,不予以缺氧损伤处理。抑制剂组加入含有JNK通路特异性阻断剂SP600125(10、20 μmol/L,溶于DMSO中)的DMEM培养基5 mL培养30 min后与缺氧组、DMSO组(加入含有与抑制剂等体积的DMSO的DMEM培养基5 mL)移入真空缺氧罐中进行缺氧性损伤处理[7],分别于缺氧处理6、12、24 h后均复氧30 min,收集细胞进行分子生物学指标检测。
1.3 α-SMA mRNA表达检测采用RT-PCR法。按Trizol RNA提取试剂盒说明提取各组的总RNA,为减少误差,每组重复3次。按反转录试剂盒的说明要求选择符合条件的mRNA进行反转录。β-actin为内参进行RT-PCR,α-SMA上游引物:5′-CTCTTCCAGCCATCTTTCATT-3′,下游引物:5′-GCATTTGCGGTGGACAATGGA-3′,片段长度:342 bp。 β-actin 上 游 引 物 :5′-GGAGATTACTGCCCTGGCTCCTA-3′,下游引物:5′-GACTCATCGTACTCCTGCTTGCTG-3′,片段长度:124 bp。反应总体系20.0 μL:上、下游引物各0.8 μL,Mix 9 μL,模板 1.5 μL,ddH2O 7.9 μL。反应条件:95 ℃ 10 min,循环1次,95 ℃ 5 s,60 ℃ 30 s,72 ℃ 40 s,循环40次。每个样本设3个副孔,取其平均值为样本Ct值。以正常组的基因表达量为1,采用2-△△Ct法计算其余各组mRNA的相对表达量。变化倍数(Fold change)为各组较正常组基因的相对表达倍数。
1.4 JNK、pJNK、α-SMA蛋白表达检测采用Western blotting法。模型建立后,按放射免疫沉淀法(RIPA)蛋白裂解液说明书抽提各组RTEC总蛋白,用二喹啉甲酸(BCA)蛋白定量分析法测定蛋白浓度。煮沸变性后取30 μg上样,进行Western Blotting。常温下十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE),湿转法转膜至聚偏二氟乙烯膜(PVDF),5%脱脂牛奶室温摇床封闭1 h,一抗JNK抗体(稀释度1∶1 000)、pJNK抗体(稀释度1∶1 000)、α-SMA抗体(稀释度1∶1 000)4℃孵育过夜,辣根过氧化物酶标记的二抗(稀释度1∶2 000)常温孵育1.5 h,利用化学发光液在FluorChem M下,检测蛋白条带灰度值,以β-actin基因蛋白作为参照,以目的蛋白/内参蛋白作为蛋白的相对表达量,采用Alpha View软件对结果进行分析。
1.5 统计学方法采用SPSS 17.0软件进行数据分析。计量资料用均数±标准差表示,多组间的比较采用单因素方差分析,两两比较采用SNK法。相关性分析采用Pearson相关分析。P<0.05为差异有统计学意义。
2 结果
2.1 各组RTEC的 α-SMA mRNA表达变化与正常组比较,缺氧 6、12、24 h组 RTEC 的 α-SMA mRNA表达均显著增加(均P<0.05),缺氧时间越长,α-SMA的mRNA表达越显著;与缺氧组相比,DMSO组RTEC的α-SMA mRNA表达改变差异均无显著性(均P> 0.05),抑制剂组(10、20 μmol/L)RTEC的α-SMA mRNA表达均显著降低(均P<0.05)。见图1。
2.2 各组RTEC的JNK、pJNK和α-SMA蛋白表达变化与正常组比较,缺氧6、12、24 h组RTEC的pJNK、α-SMA蛋白表达均显著增加(P<0.05),缺氧时间越长,JNK、pJNK和α-SMA蛋白的表达越显著;与缺氧组相比,DMSO组RTEC的JNK、pJNK、α-SMA蛋白表达差异均无显著性(均P>0.05),抑制剂组(10、20 μmol/L)RTEC的pJNK、α-SMA蛋白表达均显著降低(均P<0.05),JNK蛋白表达均显著增加(均P<0.05)。见图2。

图1 各组细胞α-SMA mRNA表达数据水平Fig.1 Analysis of alpha-SMA expression in each group
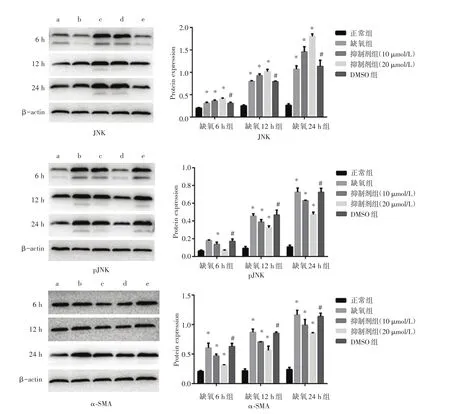
图2 各组细胞JNK、pJNK和α-SMA蛋白条带图及数据分析Fig.2 Banding patterns and data analysis of JNK,pJNK and α-SMA in each group of cells
2.3 缺氧性RTEC损伤中pJNK蛋白表达与α-SMA蛋白表达的相关性分析在缺氧性RTEC损伤中,pJNK蛋白表达与α-SMA蛋白表达呈显著正相关(r=0.950,P< 0.01)。
3 讨论
RIF是导致多种急、慢性肾病终末期的主要原因和共同病理过程[8],主要病理变化是细胞外基质的代谢失衡。本课题组的前期研究显示,在RIF中肾脏组织的α-SMA呈现高表达[9]。许春杰等[10]报道,肝纤维化中α-SMA出现高表达。缺血缺氧应激状态下,肾小管上皮细胞为适应微环境的改变,发生细胞表型的转分化,由上皮细胞转分化为肌成纤维细胞,同时加重局部炎症反应,增加致纤维化因子的表达,α-SMA则是这种表型转分化的标志蛋白[11],转分化形成的肌成纤维细胞不断增加引起的细胞外基质沉积、代谢紊乱是引起RIF的关键因素之一[12]。因此,研究肾小管上皮细胞转分化的机制对防治RIF具有重要意义。
JNK是一种可以有效磷酸化c-Jun氨基末端的唯一的蛋白激酶,又叫应激激活蛋白激酶(SAPKs),属于进化上保守的丝氨酸/苏氨酸蛋白激酶,是MAPKs超家族成员之一[13]。JNK通路可被多种应激刺激(如紫外线、高渗、缺血缺氧等)、细胞因子、生长因子以及某些G蛋白偶联受体激活,其基因可以编码46和55 kD两种蛋白产物,通过选择性剪接形成10种剪接变体[14],在不同组织发挥不同的功能及作用。有研究表明,JNK通路与组织纤维化中细胞的转分化作用有密切联系。ZHAO等[15]报道,在成纤维细胞转分化的过程中,JNK在其中起到调控作用。KLUWE等[16]认为,JNK参与了肝纤维化中转分化过程和纤维生成,其可能成为抗纤维化治疗的潜在靶标,以上研究提示JNK参与了组织纤维化中的转分化过程。
本研究通过建立缺氧性RTEC损伤模型,于缺氧后6、12、24 h分别检测细胞的α-SMA的mRNA表达,JNK、pJNK和α-SMA的蛋白表达。结果显示,缺氧使细胞的pJNK蛋白表达显著增加,缺氧时间越长,活化JNK蛋白表达越高,同时α-SMA m-RNA和蛋白表达也显著增加,高α-SMA表达提示RTEC表型转分化不断加重,提示在缺氧条件下,缺氧时间越长,缺氧诱导磷酸化的JNK越多,通路激活程度越高。相关性分析显示RTEC中的pJNK蛋白表达与α-SMA蛋白表达呈显著正相关。加入JNK通路特异性阻断剂后细胞的JNK蛋白表达增加,pJNK蛋白表达均显著减少,pJNK蛋白表达/JNK蛋白表达减少。抑制剂浓度增加时,JNK蛋白表达增加越显著,pJNK蛋白表达和pJNK蛋白表达/JNK蛋白表达减少越显著,同时抑制JNK通路后,α-SMA的m-RNA和蛋白表达均显著降低,提示抑制JNK通路的活化后,减少RTEC发生表型转分化,因此α-SMA的表达减少,JNK通路被抑制的程度越高,α-SMA的表达越低。与缺氧组相比,DMSO组各项指标均没有显著差异,提示用于溶解SP600125的DMSO并没有参与RTEC缺氧性损伤。综上所述,在大鼠RTEC缺氧性损伤中,JNK通路可能参与了RTEC表型转分化作用。本研究虽然初步讨论了JNK通路在缺氧性RTEC转分化中的作用,但并未能明确其具体分子机制,同时与JNK通路相关的其他通路在RTEC转分化中的作用如何,仍需进一步的研究证实。
[1]ZHOU T B,OU C,RONG L,et al.Effect of all-trans retinoic acid treatment on prohibitin and renin-angiotensin-aldosterone system expression in hypoxia-induced renal tubular epithelial cell injury[J].J Renin Angiotensin Aldosterone Syst,2014,15(3):243-249.
[2]LUO F,SHI J,SHI Q,et al.Mitogen-Activated Protein Kinases and Hypoxic/Ischemic Nephropathy[J].Cell Physiol Biochem,2016,39(3):1051-1067.
[3]THONGNUANJAN P,SOODVILAI S,CHATSUDTHIPONG V,et al.Fenofibrate reduces cisplatin-induced apoptosis of renal proximal tubular cells via inhibition of JNK and p38 pathways[J].J Toxicol Sci,2016,41(3):339-349.
[4]KIM C S,CHOI J S,JOO S Y,et al.Nicotine-induced apoptosis in human renal proximal tubular epithelial cells[J].PLoS one,2016,11(3):e0152591.
[5]DING Y,YANG H,XIANG W,et al.CD200R1 agonist attenuates LPS-induced inflammatory response in human renal proximal tubular epithelial cells by regulating TLR4-MyD88-TAK1-mediated NF-kappaB and MAPK pathway[J].Biochem Biophys Res Commun,2015,460(2):287-294.
[6]HONMA S,NAKAMURA K,SHINOHARA M,et al.Effect of amlodipine on mouse renal interstitial fibrosis[J].Eur J Pharmacol,2016,780(2):136-141.
[7]OUYANG F,HUANG H,ZHANG M,et al.HMGB1 induces apoptosis and EMT in association with increased autophagy following H/R injury in cardiomyocytes[J].Int J Mol Med,2016,37(3):679-689.
[8]LI A,ZHANG X,SHU M,et al.Arctigenin suppresses renal interstitial fibrosis in a rat model of obstructive nephropathy[J].Phytomedicine,2017,30:28-41.
[9]陈静,覃远汉,赵艳君,等.PAX2和α-SMA在肾小管间质纤维化大鼠肾脏中的表达及意义[J].现代预防医学,2010,37(20):3950-3952.
[10]许春杰,顾磊,蒋春晖,等.1,25(OH)_2D_3抑制肝星状细胞激活和调控Th细胞分化治疗肝纤维化[J].实用医学杂志,2017,33(13):2100-2104.
[11]ROACH K M,FEGHALI-BOSTWICK C,WULFF H,et al.Human lung myofibroblast TGFbeta1-dependent Smad2/3 signalling is Ca(2+)-dependent and regulated by KCa3.1 K(+)channels[J].Fibrogenesis Tissue Repair,2015,8:5.
[12]GREGORINI M,CORRADETTI V,ROCCA C,et al.Mesen-chymal stromal cells prevent renal fibrosis in a rat model of unilateral ureteral obstruction by suppressing the renin-angiotensin system via HuR[J].PLoS One,2016,11(2):e0148542.
[13]WESTON C R,LAMBRIGHT D G,DAVIS R J.Signal transduction.MAP kinase signaling specificity[J].Science,2002,296(5577):2345-2347.
[14]JOHNSON G L,NAKAMURA K.The c-jun kinase/stress-activated pathway:regulation,function and role in human disease[J].Biochim Biophys Acta,2007,1773(8):1341-1348.
[15]ZHAO B,GUAN H,LIU J Q,et al.Hypoxia drives the transition of human dermal fibroblasts to a myofibroblast-like phenotype via the TGF-beta1/Smad3 pathway[J].Int J Mol Med,2017,39(1):153-159.
[16]KLUWE J,PRADERE J P,GWAK G Y,et al.Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition[J].Gastroenterology,2010,138(1):347-359.
