迟缓爱德华氏菌刺激对牙鲆转录组差异基因表达的影响
2018-05-07郑津辉李庆亚耿绪云潘宝平孙金生
周 密 ,郑津辉 ,李庆亚 ,耿绪云 ,潘宝平 ,孙金生 ,,高 虹
(1.天津师范大学 生命科学学院,天津 300387;2.天津师范大学 天津市动植物抗性重点实验室,天津 300387;2.天津市水产养殖病害防治中心,天津 300221)
牙鲆(Paralichthys olivaceus)别称比目鱼,隶属于牙鲆科(Paralichthyidae)牙鲆属(Paralichthys),具有生长快速、体型庞大、肉质鲜美、营养价值高等优点,已成为亚洲海水养殖鱼类的代表之一[1].牙鲆养殖过程中,养殖环境、生长速率以及抗病能力等是影响牙鲆产量和质量的重要因素.牙鲆的疾病种类很多,其中细菌性疾病会严重危害牙鲆养殖,如鳗弧菌病、爱德华氏综合病等[2-3].迟缓爱德华氏菌(Edwardsiella tarda)属于革兰氏阴性菌,不仅能使较多的经济鱼类患病,给水产养殖业造成严重经济损失,也是一种人、鱼共患病原菌,直接威胁人类健康[4].在人工养殖环境条件下,由于养殖密度大、水体环境中代谢物及饵料沉积、抗生素药物的使用等原因,水体水质差,极易引发疾病,因此选育抗病能力强的牙鲆鱼种是推动牙鲆养殖业发展的重要途径之一[5].利用遗传学方法育种是目前牙鲆品种改良的重要手段,除了传统的杂交育种之外,对牙鲆遗传结构进行分析和改造也为培育抗病性能优良品种提供了理论依据,如Sekino等[6]和Coimbra等[7]分离了牙鲆大量的微卫星DNA,为种群遗传分析、遗传图谱的构建奠定了基础.高通量转录组测序(RNA-seq)是在转录水平上获得大量基因序列信息的一种技术,目前广泛应用于真核生物的基因测序[8-9].RNA-seq在研究基因功能、基因表达以及新基因的挖掘等方面应用越来越多,成为研究鱼类遗传学的一种重要方法[10-11].如陈阿琴等[12]对不同光周期条件下牙鲆的倒数8节尾椎骨进行转录组测序,功能富集分析表明差异基因主要富集在钙离子信号转导、光信号转导等通路上.
本研究利用迟缓爱德华氏菌刺激牙鲆,对其肾脏组织进行转录组测序和生物信息学分析,包括基因功能注释、SNP(单核苷酸多态性)分析、SSR(简单重复序列)分析等,探讨差异基因的表达情况,为研究牙鲆的基因功能、发掘新的免疫基因和克隆提供参考.
1 材料与方法
1.1 材料及处理
实验用牙鲆购于天津市滨海新区水产养殖场,选择生长良好的牙鲆6尾(免疫组、对照组各3尾,设置3个平行处理),体长(10±2)cm.为了消除环境因素对牙鲆生长的影响,选择可供氧、控温和水过滤的水循环鱼缸,并对鱼缸进行消毒.卤盐盐度为1.7%~1.8%(质量分数),水温20℃左右,充氧处理7 d.实验开始前让牙鲆在调配好的水环境下暂养1 d以适应环境.牙鲆营底栖生活,不适宜强光照射,因此对鱼缸进行遮光处理,每天喂食(人工配置饲料)1次.
迟缓爱德华氏菌来自天津市水产养殖病害防治中心,将其接种于pH值为7.2的LB液体培养基中,放入28℃恒温震荡培养箱(200 r/min)中培养24 h.当菌液OD600值等于1.0时,在4℃、12 000 r/min条件下离心10 min,收集菌体.感染前用PBS离心洗涤菌体3次,利用血球计数板计数,用PBS将菌液稀释到1×107CFU/mL,立即注射进免疫组牙鲆体内.
1.2 主要仪器和试剂
1.2.1 仪器
Hiseq 2000测序仪,美国Illumina公司;核酸蛋白检测仪,美国Thermo公司;PCR仪、凝胶成像仪,美国Bio-Rad公司;电泳仪,北京六一仪器厂.
1.2.2 试剂
Total RNA提取试剂盒、PCR扩增试剂盒,上海生工生物有限公司;cDNA第一链合成试剂盒,美国Sangon BiotechR公司;DNA Maker,大连宝生物科技有限公司.
1.3 方法
1.3.1 样品采集和总RNA抽提
对免疫组牙鲆注射迟缓爱德华氏菌,注射量为1.5mL;对照组牙鲆仅注射PBS缓冲液,注射量也为1.5mL.6 h后解剖鱼的肾脏,将其剪切成小块储存于冷冻管中,立即放入液氮冷冻,之后存于-80℃冰箱中用于RNA提取.由北京诺禾致源有限公司提取RNA,每组3个样品所提取的RNA等量混合,用于测序.
1.3.2 建库测序
建库测序由北京诺禾致源有限公司完成,基本流程如下:
①琼脂糖凝胶电泳检测分析后,选取未被降解、完整性好且没有被污染的RNA,使用Nanodrop微量核酸定量仪检测RNA的纯度,使用Qubit荧光定量仪测定RNA的浓度.采用Agilent 2100对RNA的质量进行检测[13-14],之后分别把每组处理中的3个样品所提取的RNA进行等量混合,用于后续实验.
②牙鲆RNA质量检测合格后,使用带有Oligo(dT)的磁珠富集 mRNA.然后用 fragmentation buffer将mRNA打断为200~700 nt(nt表示核苷酸数量)的片段,以其为模板合成第一条cDNA链,在反应体系中再加入缓冲液、DNA聚合酶I、RNase H、dNTPs以及随机引物,合成双链cDNA.用AMPure XP beads纯化回收合成的双链cDNA,进行粘性末端修复和加A尾处理,连接上测序接头后对cDNA片段进行筛选[13,15].PCR扩增后,利用AMPure XP beads纯化扩增产物,最后进行文库构建.利用Qubit2.0、Agilent 2100和QPCR方法对建好的文库进行质量检测,质检合格后,采用Illumina HiseqTM2000完成转录组测序[13,16].
1.3.3 序列拼接和组装
对原始测序数据(raw reads)进行过滤,去除带有接头的序列、低质量序列(质量值Qphred≤20的碱基数占整个序列50%以上的序列)和不确定的碱基信息占整条序列10%的序列,得到clean reads.利用转录组拼接软件Trinity对clean reads进行拼接形成转录本,利用Corest对拼接得到的转录本进行层次聚类,选取最长的Cluster序列进行功能注释分析[17].
1.3.4 转录本功能的注释
利用生物信息学方法对拼接好的Unigene的基因结构、表达水平和基因功能进行注释.为了获取更全面的基因功能信息,进行多个数据库的比对,包括蛋白数据库 NR(NCBI non-redundant protein sequences)、NT(NCBI nucleotide sequences)、PFAM(Protein family)、KOG/COG(clusters of orthologous groups of proteins/euKaryotic ortholog groups)、Swiss-Prot(a manually annotated and reviewed protein sequence database)、KEGG(Kyoto encyclopedia of genes and genomes)、GO(gene ontology)[14,18].通常用 blastx 的方式将 Unigene序列分别和NR、Swiss-Prot、KEGG和COG数据库进行比对,从而得到给定Unigene的蛋白功能注释信息.与NR数据库进行比对后,可获取牙鲆基因序列与其近缘物种的相似性,并且对牙鲆基因进行功能注释[14];依据NR等数据库的注释信息,对牙鲆基因进行GO功能注释和分类,可以从宏观水平上了解牙鲆基因的分布情况[13];依据COG数据库能够对基因产物进行同源性分类[13];目的基因和KEGG信号通路数据库比对之后,可以对目的基因所参与的信号通路进行注释,从而确定目的基因在其信号通路中的位置和功能,为确定基因的相关生物学途径提供理论依据[13].
1.3.5 差异表达基因(DEGs)的筛选和分析
差异表达基因分析是为了找出免疫组和对照组相比表达发生变化的基因.比较牙鲆对照组与免疫组的基因表达水平,使用RSEM软件对比对结果进行定量分析,采用FPKM方法对定量分析结果进行基因表达量的估算.采用TMM对read count标准化处理后,利用DESeq算法进行差异表达分析,以q<0.005且|log2Fold Change|>1为筛选差异基因功能的标准.筛选出迟缓爱德华氏菌刺激6 h后牙鲆肾脏的差异显著表达基因,对差异基因进行GO功能富集分析,获得GO term被差异基因富集的概率,着重分析显著富集的GO term,对其中的基因总数进行统计.为了研究基因的功能,对每一个组合上调和下调的差异基因进行KEGG通路富集分析,从而对差异基因所参与的生化代谢途径和信号转导通路进行注释.利用下列分析计算公式筛选出差异基因显著富集的通路(pathway),并统计出富集通路中差异基因的数量,以P<0.05视为显著富集.

式中:N表示所有的通路数目;M表示所有注释为某一通路的基因数;n表示在N中的差异表达基因数;m表示某一通路中差异表达的基因数.
1.3.6 差异表达基因验证
生物信息学分析因为组装和数据处理的问题常会产生误差,为了验证本次转录组测序的组装结果和差异基因筛选的准确性,选择4个转录组中提示的4个差异表达基因(IL-21,白介素-21;IL-1,白介素-1;IL-15,白介素-15;TNFα,肿瘤坏死因子-α),采用荧光定量PCR方法,分析4个基因在牙鲆注射迟缓爱德华氏菌6 h后的相对表达情况.实验材料见第1.2.1部分,利用Trizol法提取样品总RNA,电泳检测RNA的质量,使用Sangon BiotechR公司的M-MuLV第一链cDNA合成试剂盒,参照说明书合成cDNA.以牙鲆β-actin基因为内参,设计4个基因的引物,如表1所示.反转录cDNA为模板,利用ABI 7500荧光PCR仪做基因表达定量分析.

表1 4个差异表达基因和β-actin基因的实时定量表达引物序列Tab.1 Real-time quantitative expression of four differentially expressed genes and β-actin gene
2 结果与分析
2.1 数据预处理
采用Illumina HiSeqTM测序技术获得了牙鲆免疫组和对照组2个转录组的数据,对原始数据进行统计,对照组获得了105 228 444条raw reads,过滤掉含有接头、低质量的reads后获得了98 735 284条clean reads,共14.81 G,GC含量为48.87%;免疫组共获得了97 358 862条raw reads,其中 clean reads有90 362 310条,共13.55 G,GC含量为48.59%.
2.2 序列拼接
用Trinity软件对clean reads进行拼接后,共得到222 980条转录本,99 808个基因.其中,转录本长度在 200~500 bp间的有 131 634条,500~1 000 bp间的42 702条,1 000~2 000 bp间的 23 275条,>2 000 bp的有25369条;基因长度在200~500bp间的有21138个,500~1 000 bp间的 30 211个,1 000~2 000 bp间的23 091个,>2 000 bp的有25 368个.转录本总长度为200 234 420 bp,长度范围为 201~22 935 bp,平均长度为898 bp,中等长度为411 bp,N50(转录本长度相加超过总长度50%时的最后一条转录本的长度)为1 886 bp,N90(转录本长度相加超过总长度90%时的最后一条转录本的长度)为319 bp.基因总长度为158 960 029 bp,长度范围为 201~22 935 bp,平均长度为1593bp,中等长度为966bp,N50(基因长度相加超过总长度50%时的最后一个基因的长度)为2633 bp,N90(基因长度相加超过总长度90%时的最后一个基因的长度)为694 bp.
2.3 基因功能注释
对组装得到的牙鲆转录组进行功能注释,结果如表2所示.
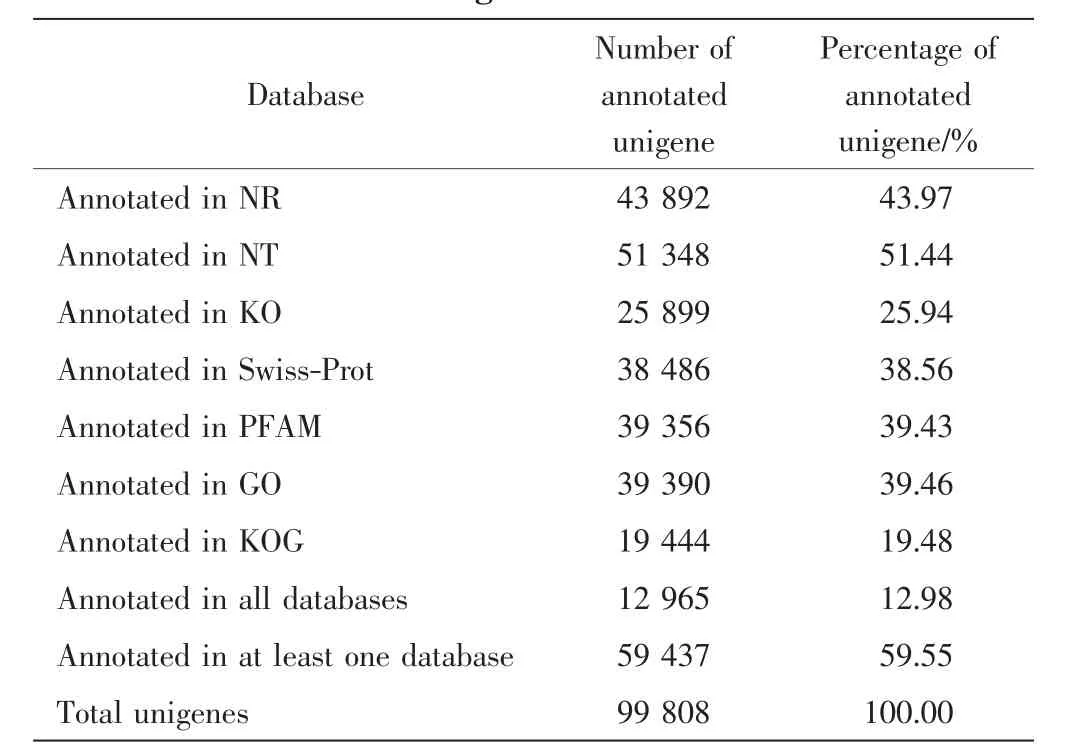
表2 基因注释成功率统计Tab.2 Statistics of gene annotation success rate
由表2可知,有51 348条Unigenes注释到NT数据库,注释率最高(51.44%);其次是NR数据库,注释了 43 839条(43.97%);PFAM、Swiss-Prot、GO 数据库的注释率相近,约为39%;KO和KOG数据库的注释率最低.
2.3.1 NR注释分析
在NR数据库中有43 892条Unigenes得到注释,分别对其进行相似性分布、E值分布(E值反映可信度的高低,E值越小代表可信度越高)和物种分布情况的分析,结果如图1所示.

图1 NR数据库比对结果的统计Fig.1 Statistics of NR database comparison results
由图1(a)可以看出,注释到的 Unigenes约有66%与NR数据库中相应蛋白的相似度超过80%,因此转录组所测得数据的可信度比较高.图1(b)的E值分布图中,NR所注释到的E值小于1×10-15的Uingenes占94.1%;在图1(c)的物种分布图中,注释到的Unigenes与大黄鱼(Larimichthys crocea)的匹配度最高,33.5%的Unigenes比对到大黄鱼中的相应蛋白,说明牙鲆与大黄鱼的亲缘性比较高;其次是深裂眶锯雀鲷(Stegastes partitus),牙鲆有28.7%的Unigenes比对到深裂眶锯雀鲷中;有24.5%的Unigenes比对到其他鱼中.
2.3.2 GO分析
对牙鲆转录组进行GO分析,结果如图2所示.牙鲆转录组一共有39 390个Unigenes注释到GO数据库中,分为生物过程、细胞组分和分子功能3类.由图2可以看出,在生物过程中注释到的基因主要分布在代谢过程、单一生物过程、细胞过程及生物调节过程中;在细胞组分中主要分布在细胞和细胞器部分;分子功能中主要分布在粘合与催化活性方面.
2.3.3 KOG功能注释分析
牙鲆转录组中KOG功能注释的Unigenes共有19 444条,注释到26个功能类别上,注释比例为19.48%,如图3所示.由图3可以可以看出,大部分Unigenes都被注释到KOG分类中的信号转导机制、一般功能预测、翻译后修饰、蛋白质周转和分子伴侣、转录、细胞内运输、分泌和水泡运输、细胞骨架、RNA加工和修饰等功能类别.
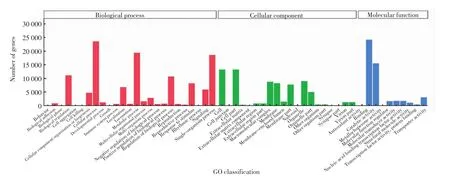
图2 GO分类Fig.2 GO classification
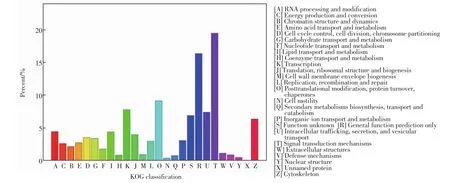
图3 KOG分类Fig.3 KOG classification
2.3.4 KEGG分类
牙鲆转录组在数据库中进行KO注释后可进一步得到所在的通路注释.本转录组所获得的序列中有25 899条能在KO数据库中得到注释,参与到232条相应的通路上.将注释到的基因所参与的KEGG代谢通路进行归类,大致有5个分支,分别是细胞过程(CP,cellular processes)、环境信息处理(EIP,environmental information processing)、遗传信息处理(GP,genetic information processing)、代谢(M,metabolism)和有机系统(OS,organismal systems),每个分支的KEGG分类结果如图4所示.免疫组牙鲆经过迟缓爱德华氏菌刺激后,同对照组相比,环境信息处理分支下的信号转导(signal transduction)注释了4 513个基因,在有机系统分支下的免疫系统(immune system)注释了1 922个基因,这为爱德华氏综合征的防治提供了新的依据.
2.4 差异基因的分析
2.4.1 差异基因的筛选
本研究共得到25 241个表达量不同的Unigenes,按照q<0.005且|log2(foldchange)|>1的原则筛选后,2 502个Unigenes视为存在显著差异表达.以对照组为参照,存在差异表达的Unigenes在免疫组中有1 280个上调,1 222个下调.免疫组和对照组均有表达的基因有51 478个,免疫组和对照组特异性表达的基因个数分别是17 235个和16 694个.
2.4.2 差异基因GO富集分析
通过GOseq方法对差异表达基因进行GO富集分析,注释到的差异表达基因有2 048个.校正后的P<0.05时,视该功能为富集项,差异基因显著富集在38个GO terms上,包括6个细胞组分、12个生物学过程和20个分子功能,如图5所示.对图5中的差异基因进行GO富集,差异基因主要富集在生物过程(BP)、细胞组分(CC)和分子功能(MF)3大类别上.差异基因的上调和下调情况如图6所示.在图6中,上调和下调的GO terms主要集中在生物学过程中的生物调节过程,包括细胞通讯、信号传导、单细胞信号转导、细胞组分中的膜的整体成分和膜的固有成分等.

图4 KEGG分类Fig.4 KEGG classification

图5 差异基因GO富集Fig.5 Differential gene GO enrichment
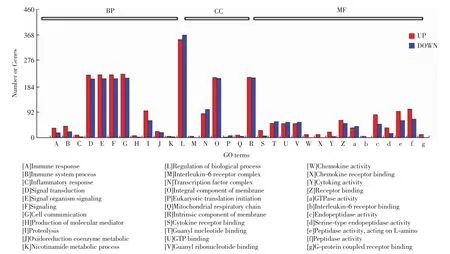
图6 差异基因的表达变化Fig.6 Differential gene expression changes
2.4.3 差异基因的KEEG富集分析
通过Rich factor、q值和富集到此通路上的基因个数来衡量KEGG的富集程度.差异基因共获得277个KEGG通路富集,其中627个差异基因显著富集到20条信号通路上.20条信号通路分别是Osteoclast differentiation、NF-kappa B signaling pathway、Measles、Cytokine-cytokine receptor interaction、Influenza A、TNF signaling pathway、Malaria、Hematopoietic cell lineage、Jak-STAT signaling pathway、Leishmaniasis、Hepatitis C、NOD-like receptor signaling pathway、Herpes simplex infection、Asthma、RIG-I-like receptor signaling pathway、Cytosolic DNA-sensing pathway、Adipocytokine signaling pathway、Legionellosis、AGE-RAGE signaling pathway in diabetic complications、Rheumatoid arthritis,其中 8 个与信号通路有关,3个与发育有关,9个与相关疾病有关.下调基因显著富集到20个KEGG通路,其中多达7条与代谢和发育有关,包括beta-Alanine metabolism、Osteoclast differentiation、Histidine metabolism、Transcriptional misregulation in cancer、Pyruvate metabolism、Phosphatidylinositol signaling system、Apoptosis-multiple species.上调基因显著富集到20个KEGG通路,有7条与信号通路有关,包括NF-kappa B signaling pathway、TNF signaling pathway、NOD-like receptor signaling pathway、Cytosolic DNA-sensing pathway、Adipocytokine signaling pathway、RIG-I-like receptor signaling pathway、Jak-STAT signaling pathway.
2.4.4 实时定量PCR结果和转录组分析结果比较
牙鲆感染迟缓爱德华氏菌6 h后,利用实时荧光定量PCR方法,分析白介素21基因(IL-21)、白介素1基因(IL-1)、白介素 15基因(IL-15)和肿瘤坏死因子-a基因(TNFα)的相对表达情况,结果如表3所示.

表3 转录组数据的Real-time PCR验证Tab.3 Real-time PCR validation of transcriptome data
由表3可知,基因的实时荧光定量PCR变化趋势与转录组结果变化趋势一致,在免疫刺激6 h后,4个基因都表现为上调趋势,说明转录组测序结果可靠.
3 讨论与结论
本研究用迟缓爱德华氏菌对牙鲆进行免疫刺激,采用Illumina HiSeq测序平台对牙鲆转录组进行测序,对照组获得了98 735 284条clean reads,共14.81 G,GC含量为48.87%;免疫组获得了90 362 310条clean reads,共13.55 G,GC含量为48.59%.经过对数据进行处理和组装后,在NR数据库中进行注释.所获得的转录组共包含222 980条转录本,其中99 808条序列得到了注释.分析注释结果发现,有94.1%的Unigenes E值小于1×10-15,同时有66%的注释Unigenes与NR数据库中相应蛋白相似度大于80%,说明大部分数据的可信度高,而且与大黄鱼的同源性很高,说明本次测序结果的质量可靠,数据覆盖率也比较广.
用迟缓爱德华氏菌对牙鲆进行免疫刺激后共筛选出2 502个差异表达基因,其中1 280个上调,1 222个下调.对差异表达基因分别进行GO和KECC富集分析,筛选出显著富集的GO terms和KEGG通路富集.对其进行分析,可以确定差异表达基因的功能以及它们所参与的信号通路.对KEGG富集分析发现,牙鲆表达量上调基因的显著富集主要与信号通路有关,迟缓爱德华氏菌刺激后,NF-κB信号通路中TNFα、IL-1β等免疫因子表达量上调,从而诱导多种免疫基因表达,参与炎症反应;其余信号通路是TNF信号通路、NOD样受体信号通路、RIG-I样受体信号通路、Jak-STAT信号通路等,其中Jak-STAT信号通路中IL-6、SHP2(非跨模型蛋白酪氨酸磷酸酶)、原癌基因(Pim-1)表达上调,该通路参与细胞的免疫调节以及分裂分化等生物学过程.根据这些信号通路中所提供的差异基因表达量变化的信息,可以为研究牙鲆应对迟缓爱德华氏菌刺激时信号通路中各基因所起作用以及筛选免疫相关基因提供理论依据[19].
参考文献:
[1]刘峰,陈松林,王磊,等.不同牙鲆群体遗传力和育种值分析[J].中国水产科学,2013,20(4):691-697.LIU F,CHEN S L,WANG L,et al.Analysis of heritability and breeding value of different stocks for Japanese flounder(Paralichthys olivaceus)[J].Journal of Fishery Sciences of China,2013,20(4):691-697(in Chinese).
[2]吴恋,孙金生,耿绪云,等.牙鲆Toll样受体1基因全长cDNA的克隆及特征分析[J].安徽农业科学,2013,40(26):12754-12760.WU L,SUN J S,GENG X Y,et al.Molecular cloning and expression of toll-like receptors 1 cDNA in Japanese Flounder,Paralichthys oli-vaceus[J].Anhui Agricultural Sciences,2013,40(26):12754-12760(in Chinese).
[3]GAO H,WU L,SUN J S,et al.Molecular characterization and expression analysis of Toll-like receptor 21 cDNA from Paralichthys olivaceus[J].Fish&Shellfish Immunology,2013,35(4):1138-1145.
[4]敖弟书,吴中明,王玉,等.迟钝爱德华氏菌感染大鲵的研究[J].四川动物,2010,29(3):411-414.AO D S,WU Z M,WANG Y,et al.Study on infection in Andrias davidianus caused by Edwardsiella tarda[J].Sichuan Journal of Zoology,2010,29(3):411-414.
[5]ANSORGE W J.Next-generation DNA sequencing techniques[J].New Biotechnol,2009,25(4):195-203.
[6]SEKINO M,HRARA M,TANIGUCHI N.Loss of microsatellite and mitochondrial DNA variation in hatchery strains of Japanese flounder Paralichthys olivaceus[J].Aquaculture,2002,213(1):101-122.
[7]COIMBRA M R M,KOBAYSSHI K,KORETSUGU S,et al.A genetic linkage map of the Japanese flounder,Paralichthys olivaceus[J].Aquachulture,2003,220(1/2/3/4):203-218.
[8]李少波,傅国辉.转录组测序数据中cSNP和表达差异基因的分析方法[J].上海交通大学学报,2014,34(2):129-133.LI S B,FU G H.RNA-Seq based analysis on cSNP and gene expression level[J].Journal of Shanghai Jiaotong University,2014,34(2):129-133(in Chinese).
[9]宋文涛,张潇峮,廖小林,等.牙鲆微卫星标记遗传连锁图谱的构建[J].农业生物技术学报,2011,19(6):981-987.SONG W T,ZHANG X Q,LIAO X L,et al.Construction of a microsatellite-based genetic linkage map in Japanese flounder(Paralichthys olivaceus)[J].Journal of Agricultural Biotechnology,2011,19(6):981-987(in Chinese).
[10]袁静.尼罗罗非鱼性腺发育过程的转录组学研究及家族生物信息学分析[D].重庆:西南大学,2013.YUN J.Characterization of Gonadal Transcriptomes from Nile tilapia{Oreochromis niloticus)and Bioinformatic Analysis of Fox Gene Family[D].Chongqing:Southwest University,2013(in Chinese).
[11]XU Q Q,LI R G,MONTE M M,et al.Sequence and expression analysis of rainbow trout CXCR2,CXCR3a and CXCR3b aids interpretation of lineage-specific conversion,loss and expansion of these receptors during vertebrate evolution[J].Developmental&Comparative Immunology,2014,45(2):201-213.
[12]陈阿琴,张影,陈松林,等.不同光周期条件下日本牙鲆尾部神经分泌系统转录组分析[J].水产学报,2016,40(6):833-843.CHEN A Q,ZHANG Y,CHEN S L,et al.The transcriptome sequencing and functional analysis of CNSS in Paralichthys olivaceus treated with different photoperiods[J].Journal of Fisheries Science,2016,40(6):833-843(in Chinese).
[13]许宝红.感病草鱼脾脏的比较转录组分析[D].长沙:湖南农业大学,2012.XU B H.Transcriptome Analysis of Grass Carp Infected GCHV[D].Changsha:Hunan Agricultural University,2012(in Chinese).
[14]北京诺禾致源科技股份有限公司.诺禾致源真核无参转录组生物信息分析结题报告[R].北京:北京诺禾致源科技股份有限公司,2013.Beijing Nuowui Source of Science and Technology Co Ltd.A Report on the Analysis of Bioinformatics in Non-transcriptome of Nucleus of[R].Beijing:Beijing Nuowui Source of Science and Technology Co.,Ltd,2013(in Chinese).
[15]COCK P J A,FIELDS C J,GOTO N,et al.The Sanger FASTQ file format for sequences with quality scores,and the Solexa/Illumina FASTQvariants[J].NucleicAcidsResearch,2010,38(6):1767-1771.
[16]HANSENKD,BRENNERS E,DUDOIT S.Biases in illumina transcriptome sequencing caused by random hexamer priming[J].Nucleic Acids Research,2010,38(12):e131.
[17]GRABHERR M G,HAAS B J,YASSOUR M,et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J].Nature Biotechnology,2011,29(7):644-652.
[18]FINN R D,TATE J,MISTRY J,et al.The Pfam protein families database[J].Nucleic Acids Research,2008,36(1):281-288.
