T细胞来源的细胞外囊泡通过miR-193a诱导足细胞损伤
2018-05-05张思盼张昌明吴俊男徐孝东朱小东刘志红
张思盼 张昌明 吴俊男 赵 越 徐孝东 郎 月 朱小东 刘志红
足细胞损伤是特发性局灶节段性肾小球硬化(FSGS)发病的中心环节[1]。研究发现T细胞功能异常参与了FSGS的发病过程,其机制可能为T细胞分泌某种循环因子导致足细胞损伤[2]。
细胞外囊泡(extracellular vesicles,EVs)是细胞间通信的重要信使,EVs包含的miRNA可在细胞间运输并引起靶细胞效应[3]。T细胞等免疫细胞是循环中EVs的重要来源[4-5]。研究显示体内足细胞可摄入白细胞来源EVs[6], 提示EVs能够进入足细胞,但循环中EVs是否对损伤足细胞具有损伤效应尚不明确。
特发性FSGS患者临床表现大量蛋白尿,病理特点为足细胞损伤及局灶节段性肾小球硬化,肾小球无炎细胞浸润或免疫复合物沉积。为了探讨FSGS患者循环中EVs的致病作用及其来源,本研究从分析FSGS患者循环EVs入手,观察其导致蛋白尿及足细胞损伤的作用,继而对其所包含miRNAs的成分进行分析,并对其是否来源于T细胞进行了研究和阐述。
对象和方法
研究对象经国家肾脏病临床医学研究中心肾活检确诊FSGS患者26例,尿蛋白定量>3.5 g/24h,血清白蛋白<30 g/L。排除家族史阳性、肥胖、HIV感染、丙型肝炎、毒物或药物(如海洛因、二膦酸盐)导致的继发性FSGS。同时入组年龄、性别相匹配的健康志愿者15例。所有入组人员均已签署知情同意书。人类样品使用按照金陵医院伦理委员会规定进行。
主要实验材料和试剂Hsa-miRNA-193a mimic(锐博生物科技有限公司)。罗丹明标记的鬼笔环肽(Cytoskeleton公司),抗Wilms瘤抑癌基因(WT1)抗体(Abcam公司),HRP标记的山羊抗兔IgG(Engene公司),化学发光试剂ECL(millipore公司),激光共聚焦显微镜(Zeiss LSM 710)。
血标本采集和血浆分离静脉采集受试者外周血40 ml,EDTA抗凝。外周血采集后6h内以3 000g离心10 min,分离血浆。
EVs分离血浆或细胞培养上清按照梯度离心法分离EVs,具体方法见参考文献[7]。主要步骤为:1、4℃ 2 000g离心10 min,取上清,去除细胞和细胞碎片;2、4℃ 10 000g 离心30 min,取上清,去除大蛋白和细胞凋亡小体等;3、4℃ 100 000g (Beckman Coulter Optima L-80 ultracentrifuge)离心60 min,弃上清,PBS洗涤沉淀;4、4℃ 100 000g (Beckman Coulter Optima L-80 ultracentrifuge)离心60 min,弃上清,50 μl PBS重悬沉淀。BCA法(碧云天)测定EVs蛋白含量。
MiRNA芯片和RT-PCR 按照说明书使用mirVana miRNA 提取试剂盒(Ambion公司)提取血浆EVs和Jurkat T细胞EVs的总RNA,由上海伯豪生物技术有限公司进行分析,抽提所得总RNA经Agilent Bioanalyzer 2100 (Agilent technologies,Santa Clara,CA,US)电泳质检合格后备用。4例FSGS患者血浆EVs和4例对照血浆EVs RNA用Agilent Human miRNA (8*60K) V19.0 芯片(design ID: 46064)测定差异表达的miRNA。扩大样本量筛选时,RNA 样品用 qRT-PCR kit (Takara Bio,Otsu,Japan)测定miRNA。Taqman探针购自Takara Bio。
细胞培养和转染条件性永生化人足细胞来自M.Saleem (University of Bristol,Bristol,United Kingdom),培养条件和文献报道一致[8]。Jurkat细胞购自上海细胞所,培养条件与atcc.org说明一致。使用LipofectamineTMRNAiMAX Transfection Reagent(Invitrogen,)按照产品说明书进行细胞转染。转染后48h收集EVs,-80℃保存备用。为了阐明EVs能否介导T细胞与足细胞之间的通信进而导致足细胞损伤,用miR-193a mimics转染Jurkat T细胞使之过表达miR-193a,收集细胞培养上清中EVs,或与体外培养的足细胞孵育,观察足细胞损伤。
动物模型制备实验动物使用经金陵医院实验动物伦理委员会批准。8周龄雄性Balb/c小鼠尾静脉注射EVs,隔天一次,共注射三次,检测尿蛋白,肾脏组织学改变。EVs经RIPA裂解后行蛋白浓度测定,每只小鼠每次注射30 μg[7]。测定注射前和注射后第6天尿白蛋白/肌酐比值,将尿白蛋白/肌酐比值>200 μg/mg定义为蛋白尿。同上,收集过表达miR-193a的EVs,注射给小鼠,观察小鼠蛋白尿、足细胞损伤等。
尿蛋白测定按照生产厂家说明书使用Albuwell M andCreatinine Companion Kits (Exocell Inc.)测定小鼠尿白蛋白和肌酐。
足细胞骨架定量分析足细胞肌动蛋白用罗丹明-鬼笔环肽染色,共聚焦显微镜拍照。罗丹明染色区域转化为黑色像素后用ImageJ 软件定量分析。灰度为0(黑色)到200(白色,细胞骨架最大值)。计算平均足细胞骨架像素比值和单细胞骨架蛋白含量。
Western印迹体外培养足细胞用含磷酸酶抑制剂和蛋白酶抑制剂(Roche)的RIPA裂解。电泳转膜后用抗WT1抗体(Abcam)孵育。
统计学方法采用Graphpad Prism 6.0统计软件进行数据分析。数据以均值±标准差表示,两组数据的比较使用非配对t检验,P<0.05为差异有统计学意义。
结 果
FSGS患者循环中EVs包涵的miR-193a可导致足细胞损伤为明确FSGS患者EVs中miRNA表达情况,我们对两组EVs进行芯片检测和分析,共检出551种miRNA。与对照组EVs相比,FSGS组升高大于1.5倍的miRNA有62个(图1A)。RT-PCR验证升高的 miRNA有48个。扩大样本量,对这48个miRNA进行RT-PCR验证,发现FSGS组有6个miRNA较对照组EVs升高(图1B)。用非参数t检验比较FSGS患者和对照EVs的miRNA的CT值,差异有统计学意义。将这6个miRNA mimics分别用10 nmol/L,50 nmol/L,100 nmol/L浓度转染足细胞,发现miR-193a(50 nmol/L,100 nmol/L)转染后的足细胞,细胞骨架出现明显损伤(P<0.05)(图1C)。

图1 FSGS患者循环EVs包涵的miR-193a导致足细胞损伤A:FSGS患者循环EVs中包涵miRNAs的筛选;B:RT-PCR结果,FSGS患者EVsmiR-193a水平较健康人EVs升高;C:miR-193a(50 nM,100 nM)转染足细胞后均可引起细胞骨架损伤;FSGS:局灶节段性肾小球硬化;EVs:细胞外囊泡;FSGS EVs:FSGS患者血浆EVs;CTL EVs:健康人血浆EVs; Scram.:miR-193a的随机对照序列Scramble;*:P<0.05
FSGS患者循环EVs导致小鼠蛋白尿和足细胞损伤将FSGS患者和正常对照循环EVs经尾静脉隔天注射Balb/c小鼠。注射后第6天时注射FSGS患者循环EVs的小鼠尿白蛋白肌酐比值异常(>200 μg/mg)比例明显高于对照组(图2A,表1)。分别用两组的EVs处理体外培养足细胞,检测细胞骨架改变。FSGS患者EVs可引起细胞肌动蛋白减少、骨架结构紊乱(图2B、C)。
表1注射健康人与FSGS患者血浆EVs后小鼠蛋白尿产生情况

对照组FSGS组总数1526产生蛋白尿小鼠数08产生蛋白尿小鼠比例030.77%
FSGS:局灶节段性肾小球硬化;EVs:细胞外囊泡
源于过表达miR-193a的T细胞的EVs导致小鼠蛋白尿及足细胞损伤用过表达miR-193a的Jurkat细胞分泌的EVs注射小鼠,电镜下Jurkat细胞分泌的EVs直径在150 nm以内(图3A),过表达miR-193a的EVs与对照EVs大小无明显差异。体外实验表明两组EVs被足细胞摄取的能力无明显差异。Mimics转染组较对照序列转染组Jurkat细胞分泌的EVs中miR-193a含量明显升高(图3B)。小鼠尾静脉隔天注射两种EVs 12d后,注射过表达miR-193a EVs的小鼠出现蛋白尿(图3C),尿蛋白肌酐比增加(图3D),电镜观察发现该组小鼠足细胞足突融合(图3E)。将Jurkat细胞分泌的EVs与足细胞共同处理,4h后可见DilC16标记EVs已进入足细胞(图3F),经检测证实足细胞内miR-193a含量明显高于对照组(图3G)。miR-193a过表达EVs处理后,体外培养的足细胞出现明显的骨架紊乱(图3H)。

图2 FSGS患者循环EVs引起小鼠蛋白尿和足细胞损伤 A:FSGS患者血浆EVs可引起小鼠尿白蛋白肌酐比值升高;B:FSGS患者EVs引起足细胞骨架损伤;FSGS:局灶节段性肾小球硬化;EVs:细胞外囊泡;FSGS EVs:FSGS患者血浆EVs;CTL EVs:健康人血浆EVs;*:P<0.05

图3 源于过表达miR-193a的T细胞的EVs诱导足细胞损伤A:电镜观察Jurkat细胞分泌的EVs;B:miR-193a mimics和scramble转染的Jurkat细胞分泌的EVs RNA进行RT-PCR;C-E:小鼠尾静脉注射过表达miR-193a EVs和对照EVs(C),ELISA法测定小鼠尿蛋白/肌酐比值(D),电镜下两组小鼠足突结构(E);F:Jurkat EVs进入足细胞;G:过表达miR-193a的EVs处理足细胞RNA进行RT-PCR;H:富含miR-193a的EVs处理足细胞后肌动蛋白纤维减少;Scram.EVs:转染miR-193a对照序列scramble的Jurkat细胞分泌的EVs;miR-193a EVs:转染miR-193a mimics的Jurkat细胞分泌的EVs;*P<0.05
过表达miR-193a的T细胞分泌的EVs通过下调WT1表达导致足细胞损伤用过表达miR-193a的Jurkat细胞分泌的EVs处理足细胞,免疫印迹法检测发现,过表达miR-193a的EVs处理的足细胞WT1表达降低(图4)。
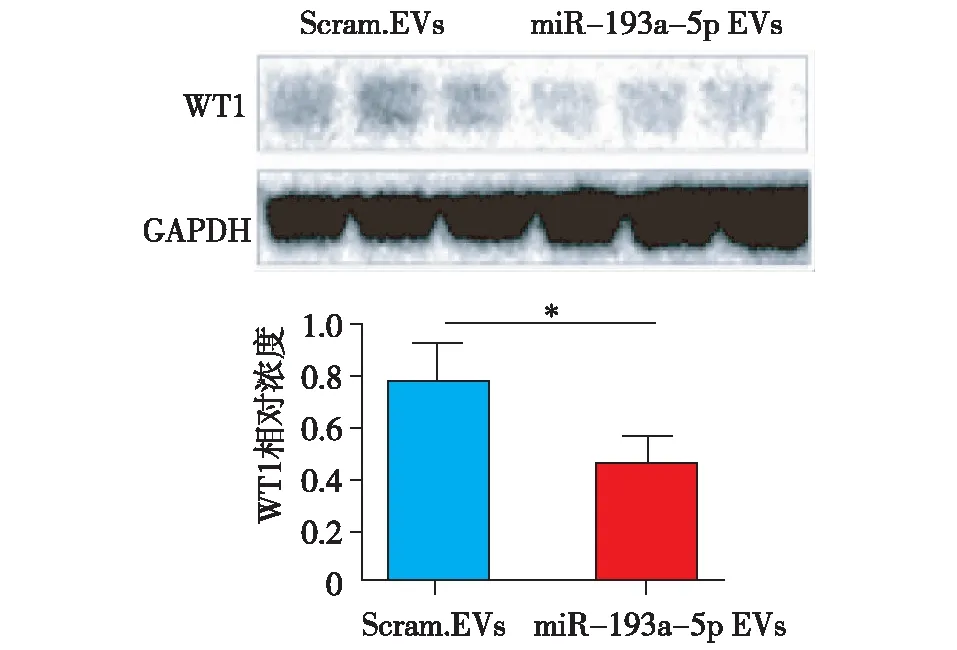
图4 富含miR-193a的EVs可下调足细胞WT1表达EVs:细胞外囊泡;Scram.EVs:转染对照序列的Jurkat细胞分泌的EVs;miR-193a EVs:转染miR-193a mimics的Jurkat细胞分泌的EVs;*P<0.05
讨 论
本研究发现FSGS患者循环中T细胞来源的EVs通过miR-193a介导足细胞损伤和蛋白尿的发生。
miRNA对维持足细胞稳态具有重要作用:损伤性miRNA可诱导足细胞损伤[9-10],例如miR-193a过表达小鼠出现蛋白尿、肾脏出现局灶节段硬化性病变[10]。近年来研究发现miRNA可通过EVs运输至靶细胞,调节靶细胞功能[11]。为了明确FSGS患者循环EVs运载的miRNA与正常人的表达差异,我们使用miRNA芯片和RT-PCR等方法筛选FSGS患者循环EVs中表达升高的miRNA,发现FSGS患者EVs中miR-193a较正常人EVs升高。体外培养足细胞过表达miR-193a后足细胞骨架结构紊乱,表明FSGS循环EVs可能通过miR-193a参与足细胞损伤。
特发性FSGS足细胞损伤严重,肾小球内无或很少有炎细胞浸润。部分FSGS患者血清能引起大鼠肾小球滤过膜通透性增加[12]。这些证据提示存在某种循环致病因子,介导足细胞损伤。EVs是细胞间通信的重要信使[13]。在EVs介导的细胞间通信中,循环EVs在远隔细胞间传递信号占重要地位[14]。循环中EVs通过肾脏排泄[15],电镜观察发现小鼠足细胞中有来自白细胞的EVs[6],说明循环EVs可到达足细胞。本文发现部分FSGS患者血浆中EVs可引起体外培养足细胞骨架损伤、小鼠蛋白尿和足细胞足突融合,表明来源于循环的EVs可介导足细胞损伤。
既往研究发现只有小部分(16.3%)特发性FSGS患者血液中存在循环致病因子[16],循环致病因子也不是特定的单一物质[2,17-19]。FSGS患者血浆注射大鼠,也只有部分大鼠产生蛋白尿[12]。此外,FSGS是一病理形态学诊断名词,病因具有多样性,病理表现不一,FSGS患者血浆EVs注射小鼠后,只有小部分小鼠产生了蛋白尿,与临床上FSGS病因多样、病理表现不同相一致。
有报道,FSGS患儿伴有T细胞功能和亚群数量的异常[20]。阿霉素肾病小鼠模型,在去除了CD4+T细胞后肾脏损伤加重[21],而去除CD8+T细胞后肾脏结构和功能都得到了保护[22]。骨髓来源的抑制性细胞通过抑制T细胞激活保护FSGS患者的足细胞[23]。因此,T细胞功能异常有可能是循环致病因子的来源之一。研究表明T细胞可通过分泌EVs调控其他细胞功能[24-25]。T细胞分泌的EVs参与类风湿关节炎等自身免疫性疾病和肝炎[26-27]。为探讨T细胞来源的EVs对足细胞的损伤作用,本文利用体外培养的T细胞系,使其过表达miR-193a,收集EVs,通过小鼠尾静脉注射和体外培养足细胞共孵育的方法,发现T细胞来源的EVs能诱导小鼠产生蛋白尿,引起足细胞足突融合和足细胞骨架结构损伤。以上结果表明T细胞来源的EVs可通过运输miR-193a导致足细胞损伤。因此T细胞来源的EVs可能是FSGS循环致病因子之一。本文未检测两组EVs的转录组学、蛋白质组学、DNA和膜脂质成分是否存在差异,曾有研究利用iTRAQ测定人为改变细胞miRNA含量后,这些细胞分泌的EVs蛋白含量,结果发现EVs的蛋白种类和表达量基本不变[28]。
张昌明等[29]发现糖皮质激素治疗后完全缓解的FSGS患者血浆miR-193a-3p明显低于大量蛋白尿的FSGS患者。Gebeshuber等[10]发现miR-193a过表达小鼠出现肾小球局灶节段硬化。miR-193a仅在特发性FSGS患者肾小球升高,而HIV相关肾病、糖尿病肾病、嘌呤霉素肾病、阿霉素肾病、suPAR模型、Heyman肾炎、硫酸鱼精蛋白灌注、Alport综合征、WT1敲除以及nephrin过表达等足细胞损伤模型中肾小球miR-193a均不升高[10],说明该miRNA具有一定的疾病特异性。过表达miR-193a转基因小鼠在8周龄时死于肾脏局灶节段硬化性病变,对不同周龄的miR-193a过表达小鼠的肾小球进行转录组学分析发现,miR-193a损伤足细胞依赖于下调WT1;WT1下调引起其靶基因podocalyxin和nephrin等对维持足细胞稳定的关键基因表达下降,由此引起整个足细胞稳定系统的毁损[10]。我们观察了过表达miR-193a的T细胞来源的EVs对足细胞的影响,发现过表达miR-193a的T细胞分泌的EVs可下调足细胞WT1表达,导致足细胞损伤。本文未直接向小鼠注射miR-193a mimics。本文未检测其他足细胞标志物变化,但是miR-193a损伤足细胞依赖于下调WT1表达,大部分足细胞功能关键基因都受WT1调控[30],仅检测蛋白表达变化不能明确是EVs中miR-193a引起的直接变化还是WT1下调引起的变化,或者EVs其他成分引起的变化。
本研究揭示了FSGS患者循环中T细胞来源的的EVs通过其运输的miR-193a诱导足细胞损伤的机制,为研究FSGS的病因提供了新的思路。
致谢:标本来自于国家肾脏疾病临床医学研究中心生物样本库。
1 Fogo AB.Causes and pathogenesis of focal segmental glomerulosclerosis.Nat Rev Nephrol,2015,11(2):76-87.
2 Maas RJ,Deegens JK,Wetzels JF.Permeability factors in idiopathic nephrotic syndrome:historical perspectives and lessons for the future.Nephrol Dial Transplant,2014,29(12):2207-2216.
3 Pegtel DM,Cosmopoulos K,Thorley-Lawson DA,et al.Functional delivery of viral miRNAs via exosomes.Proc Natl Acad Sci U S A,2010 Apr 6;107(14):6328-6333.
4 Chatila TA,Williams CB.Regulatory T cells:exosomes deliver tolerance.Immunity,2014,41(1):3-5.
5 Lv LL,Cao Y,Liu D,et al.Isolation and quantification of microRNAs from urinary exosomes/microvesicles for biomarker discovery.Int J Biol Sci,9(10):1021-1031.
6 Ståhl AL,Arvidsson I,Johansson KE,et al.A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles.PLoS Pathog,2015,11(2):e1004619.
7 Bruno S,Grange C,Deregibus MC,et al.Mesenchymal stem cell-derived microvesicles protect against acute tubular injury.J Am Soc Nephrol,2009,20(5):1053-1067.
8 Mundel P,Reiser J,Zúiga MBA,et al.Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines.Exp Cell Res,1997,236(1):248-258.
9 Henique C,Bollée G,Loyer X,et al.Genetic and pharmacological inhibition of microRNA-92a maintains podocyte cell cycle quiescence and limits crescentic glomerulonephritis.Nat Commun,2017,8(1):1829.
10 Gebeshuber CA,Kornauth C,Dong L,et al.Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1.Nat Med,2013,19(4):481-487.
11 Collino F,Bruno S,Incarnato D,et al.AKI Recovery Induced by Mesenchymal Stromal Cell-Derived Extracellular Vesicles Carrying MicroRNAs.J Am Soc Nephrol,2015,26(10):2349-2360.
12 Tanaka R,Yoshikawa N,Nakamura H,et al.Infusion of peripheral blood mononuclear cell products from nephrotic children increases albuminuria in rats.Nephron,1992,60(1):35-41.
13 Tkach M,Théry C.Communication by Extracellular Vesicles:Where We Are and Where We Need to Go.Cell,2016,164(6):1226-1232.
14 Njock MS,Cheng HS,Dang LT,et al.Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs.Blood,2015,125(20):3202-3212.
15 Lai CP,Mardini O,Ericsson M,et al.Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter.ACS Nano,2014,8(1):483-494.
16 李一鸣,赵娇,刘娟,等.MiR-18 b-5 p对大肠癌PTEN/PI3 K/Akt2信号通路的调控.现代肿瘤医学,2015,(15):2092-2096.
17 McCarthy ET,Sharma M,Savin VJ.Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis.Clin J Am Soc Nephrol,2010,5(11):2115-2121.
18 Trachtman H,Reiser J.suPAR is the circulating factor in some but not all FSGS.Nat Rev Nephrol,2014,10(10):610.
19 Kim AH,Chung JJ,Akilesh S,et al.B cell-derived IL-4 acts on podocytes to induce proteinuria and foot process effacement.JCI Insight,2017,2(21).pii:81836.
20 Hulton SA,Shah V,Byrne MR,et al.Lymphocyte subpopulations,interleukin-2 and interleukin-2 receptor expression in childhood nephrotic syndrome.Pediatr Nephrol,1994,8(2):135-139.
21 Wang Y,Wang Y,Feng X,et al.Depletion of CD4(+) T cells aggravates glomerular and interstitial injury in murine adriamycin nephropathy.Kidney Int,2001,59(3):975-984.
22 Wang Y,Wang YP,Tay YC,et al.Role of CD8(+) cells in the progression of murine adriamycin nephropathy.Kidney Int,2001,59(3):941-949.
23 Li L,Zhang T,Diao W,et al.Role of Myeloid-Derived Suppressor Cells in Glucocorticoid-Mediated Amelioration of FSGS.J Am Soc Nephrol,2015,26(9):2183-2197.
24 Blanchard N,Lankar D,Faure F,et al.TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex.J Immunol,2002,168(7):3235-3241.
25 Mittelbrunn M,Gutiérrez-Vázquez C,Villarroya-Beltri C,et al.Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells.Nat Commun,2011,2:282.
26 Distler JH,Jüngel A,Huber LC,et al.The induction of matrix metalloproteinase and cytokine expression in synovial fibroblasts stimulated with immune cell microparticles.Proc Natl Acad Sci U S A,2005,102(8):2892-2897.
27 Kornek M,Popov Y,Libermann TA,et al.Human T cell microparticles circulate in blood of hepatitis patients and induce fibrolytic activation of hepatic stellate cells.Hepatology,2011,53(1):230-242.
28 Yin Y,Cai X,Chen X,et al.Tumor-secreted miR-214 induces regulatory T cells:a major link between immune evasion and tumor growth.Cell Res,2014,24(10):1164-1180.
29 Zhang C,Zhang W,Chen HM,et al.Plasma microRNA-186 and proteinuria in focal segmental glomerulosclerosis.Am J Kidney Dis,2015,65(2):223-232.
30 Briggs PPM,Abney KD,Schoonover JD,et al.Synthesis,structure,and properties of Cs(4)Th(4)P(4)Se(26):a quaternary thorium selenophosphate containing the (P(2)Se(9))(6-) anion.Inorg Chem,2001,40(19):4871-4875.
