TCIRG1基因突变致婴儿恶性石骨症1例并文献复习
2018-04-25张潇潇吴蓓蓉董晓艳顾浩翔
张潇潇 陆 敏 吴蓓蓉 董晓艳 顾浩翔
1 病例资料
女,8月龄,因“咳嗽10 d,病程中发热1次”入住上海市儿童医院(我院)。患儿于入院前10 d与感冒家人接触后出现咳嗽,为阵发性连声咳,有痰不易咳出,病程中发热1次,热峰38.3℃,无喘息,无吐泻,至外院就诊。血常规:WBC 26.79×109·L-1,N 0.26,L 0.62,CRP 2.55 mg·L-1。X线胸片示两肺纹理增粗、增多,胸廓诸骨密度增高,两侧肱骨密度增高,近端干骺端骨质破坏,邻近见骨膜反应。先后
予头孢他啶、环酯红霉素和罗氏芬治疗,未见好转。为进一步诊治,收入我院。病程中精神、胃纳可,二便正常。
出生史及家族史:患儿系G1P1,足月顺产,出生史无异常。患儿母亲孕期有缺钙,父母非近亲结婚,否认家族性、遗传性病史。
既往史:6月龄时外院确诊先天性髋关节脱位。
查体:T 36.7℃,P 150 ·min-1,R 34·min-1,体重7 kg(P3),身长65 cm(P3),BP 82/48 mmHg。神志、精神可,呼吸平稳,面色略苍白,全身皮肤未见皮疹及淋巴结肿大。方颅,前额突出,双眼震颤,颈软,咽部充血,牙齿未萌出,漏斗胸,双侧肋缘外翻,双肺呼吸音粗,双肺可闻及痰鸣音。心率150·min-1,律齐,未及杂音。腹部膨隆,肝肋下3 cm,脾肋下3 cm,质韧。双下肢髋关节复位带固定中。肛门、生殖器未见异常,神经系统查体未见异常。
实验室检查:血常规示WBC 16.05×109·L-1,RBC 2.83×1012·L-1,Hb 78 g·L-1,PLT 141×109·L-1,N 0.26,L 0.64,CRP 5 mg·L-1,网织红细胞7.42%,有核红细胞47×109·L-1。肝肾功能电解质正常,碱性磷酸酶960 U·L-1,乳酸脱氢酶893 U·L-1,球蛋白5 g·L-1,血清钙2.03 mmol·L-1,25羟维生素D 31.68 nmol·L-1。血串联质谱未见异常。病原学检查阳性:呼吸道包括近平滑假丝酵母菌(痰培养)、军团菌(血清抗体)、鼻病毒(血清抗体)、单纯疱疹病毒(血清抗体);胃肠道包括诺如病毒(粪便病毒RNA)、光滑念珠菌(粪便培养)。骨髓穿刺细胞学:骨髓增生活跃,粒红比倒置,粒系增生,红系增生明显活跃,巨核细胞系增生低下,血小板散在少见。骨髓细胞免疫分型未见异常。
影像学及其他检查:X线片示两肺纹理增粗、增多,胸廓诸骨密度增高,两侧肱骨密度增高,近端干骺端骨质破坏,邻近见骨膜反应;双侧股骨及胫腓骨改变,考虑骨代谢性疾病(图1)。头颅MRI示脑发育不良,幕上脑室积水伴脑白质水肿,扁桃腺位置低下。眼底及听力检查未见异常。
诊治经过:入院后予阿莫西林克拉维酸钾、红霉素和伊曲康唑抗感染,丙种球蛋白支持治疗。血液科会诊,考虑婴儿恶性石骨症(IMO),不能排除合并其他遗传性疾病,建议行高通量二代基因测序明确诊断。住院10 d后患儿肺炎好转出院。
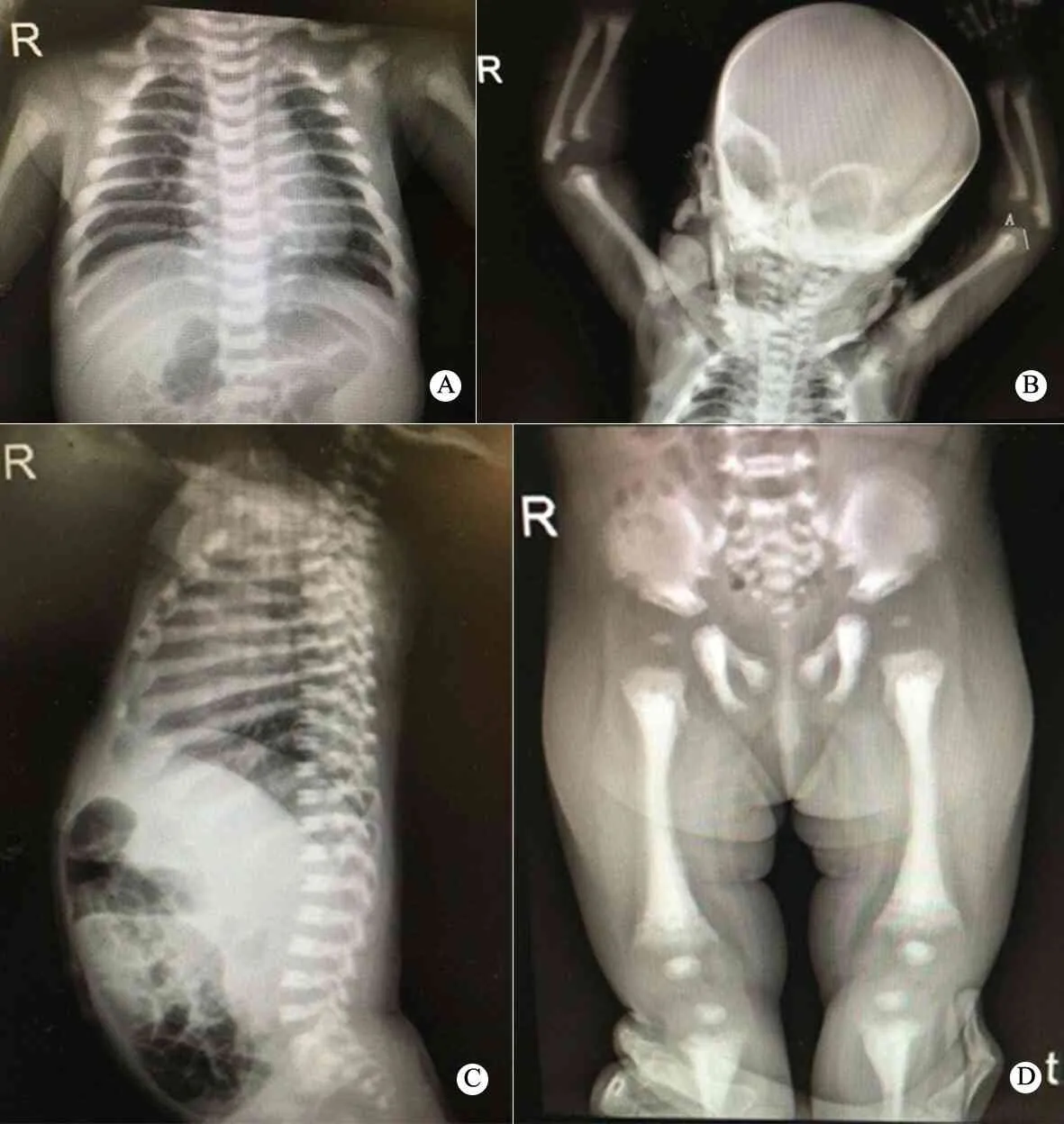
图1 患儿全身骨骼X线表现
注 A:肋骨、锁骨等胸部骨骼密度明显增高;B.颅骨明显骨化,颅底骨密度高;上肢可见“骨中骨”征象;C.椎体上下缘增厚致密,中央部密度相对较低,呈现“夹心椎”征象;D.髂骨翼呈“晕轮样”改变,下肢长骨密度明显增高,干骺端呈“烧瓶样”改变
基因检测方法及结果:采集患儿及父母外周静脉血,患儿行全基因组高通量测序(遗传性血液和免疫系统疾病基因检测)检查。血样抽提基因组DNA后,用超声仪将DNA随机打断成300~400 bp片段,磁珠纯化后产物经末端修复后进行接头连接、PCR扩增,形成测序文库,使用illumina测序仪测序(Pair End,片段长度150 bp)。采用Qubit检测文库浓度,Agilent 2100 Bio analyzer检测文库片段长度和浓度。原始测序(fastq格式)数据首先采用FastQC进行数据质量评估,给出基本统计信息、碱基质量分布、ATCG比例分布、GC含量分布、测序读长分布、重复水平评估和接头序列比例等数值;采用BWA和GATK算法推荐流程将原始数据比对到参考基因组(hg19)得到bam格式文件,运用GATK中部分模块对比对结果进行质量统计,包括比对率、PCR冗余度、测序深度等;采用GATK算法推荐流程从bam格式文件中进行变异获取并统计,包括单核苷酸位点变异 (SNV)、小片段插入和缺失、拷贝数变异 (CNV)、结构变异 (SV)。获取变异后,由上海新培晶医学检验所生物信息小组开发的遗传性血液和免疫系统疾病基因检测自动分析系统进行后续分析。该系统首先对变异位点进行筛选、注释和分析,包括数据质控、位点位置筛选、公共人群频率筛查、本地人群频率筛查、疾病数据库(如ClinVar、dbSNP、ExAc、1000 Genomes Project等)对变异进行注释、ANNOVAR功能注释、疾病遗传模式评估、公共数据库或内部数据库既往报道情况的分析等步骤,得到优选致
病位点,进一步得到符合临床表型的疑似致病位点,交由上海新培晶医学检验所分析报告团队进行人工审核。同时对有意义的突变位点进行一代测序验证。基因检测发现TCIRG1基因存在无义突变c.1360C>T(p.R454X)和移码缺失c.722delC(p.T241fs),分别遗传自患儿父母,考虑为复合杂合致病。
随访:感染情况控制良好,IMO病变未进一步恶化,目前于北京市儿童医院血液科等待配型中。
2 文献复习
在PubMed、Springer Link和Elsevier数据库中检索IMO的英文文献,以PubMed为例,检索式为(("osteopetrosis"[MeSH Terms] OR "osteopetrosis"[All Fields]) OR (Autosomal[All Fields] AND recessive[All Fields] AND ("osteopetrosis"[MeSH Terms] OR "osteopetrosis"[All Fields]))) OR (Infantile[All Fields] AND malignant[All Fields] AND ("osteopetrosis"[MeSH Terms] OR "osteopetrosis"[All Fields]))。在万方、CNKI数据库中检索国内有关IMO的报道,以万方数据库为例,检索式为cql://keywords= "石骨症" OR "骨硬化症" OR "大理石骨病"。检索时间为建库至2018年6月17日。共检出100篇相关文献[s1-s100][s1-s100文献见中国循证儿科杂志电子版(www.cjebp.net)该文附件],报道IMO 238例,根据病例来源分类,国内128例[s1-s33](包括本文1例),国外110例[s34-s100]。
77例(国外58例[s34-s61],国内19例)[s1-s11]行基因测序,共发现6种致病基因,国内19例均为TCIRG1基因突变(表1)。

表1 77例婴儿恶性石骨症患儿基因检测结果[n(%)]
表2显示已报道的IMO相关TCIRG1基因突变位点有51个(包括本例),包括缺失突变(16/51,31.4%)、剪切异常突变(10/51,19.6%)、无义突变(9/51,17.6%)、错义突变(8/51,15.7%)、插入突变(2/51,3.9%)、调控位点突变(1/51,2.0%)和其他未知突变(5/51,9.8%)。本例突变未在NCBI数据库及PubMed报道,为新发突变。
IMO主要临床特征为贫血、肝脾肿大和视力障碍,不同基因型临床特征见表3。
64例行造血干细胞移植,移植后死亡16例。死亡原因主要是移植失败(4例,25%)、移植后并发症(7例,43.7%)和感染(5例,31.3%)。

表2 TCIRG1基因突变情况
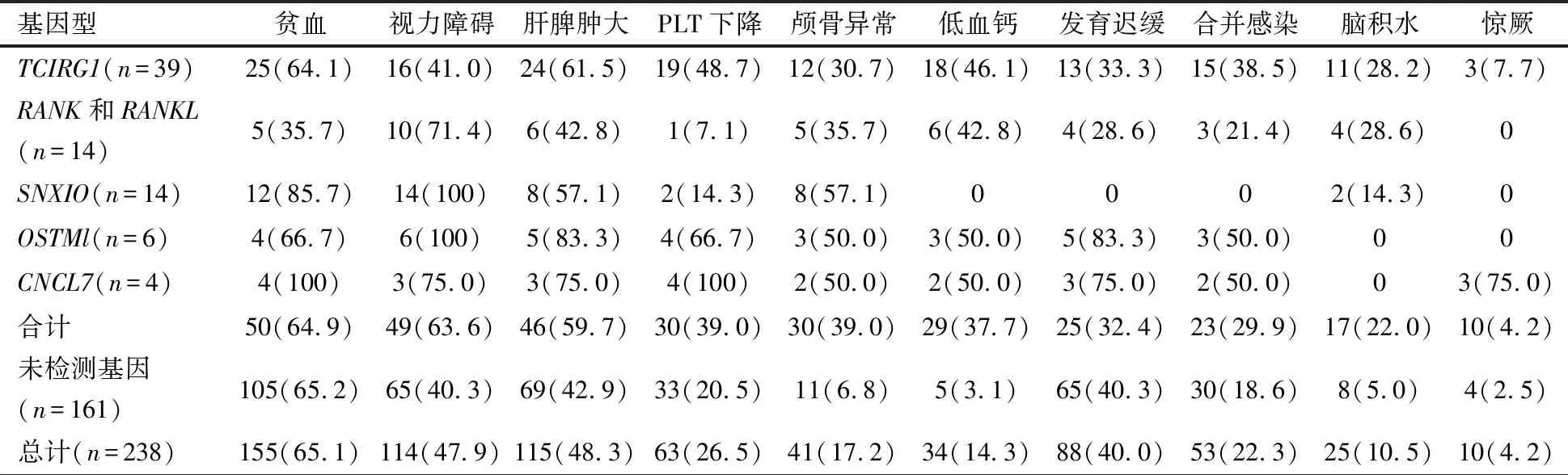
表3 婴儿恶性石骨症不同基因型的临床特征
3 讨论
IMO是一种以破骨细胞分化或功能异常为主要病变的遗传性骨代谢异常综合征[24],为常染色体隐性遗传病,发病率约为1/250 000,是石骨症中最严重的一种类型[25]。
IMO临床症状缺乏特异性,确诊依赖X线片检查[26],主要表现为全身广泛均匀的骨密度增高,髓腔狭窄或闭塞[27]。本文患儿婴儿期起病,临床特点有方颅、眼球震颤、牙齿萌出延迟、佝偻病、肝脾肿大、贫血、脑积水和脑发育不良,X线片可见广泛的骨密度增高、干骺端骨质破坏、“夹心椎”征象,符合IMO特征。

目前发现TCIRG1、CLCN7、OSTMI、RANK、RANKL和SNXIO基因参与IMO发病。IMO患者检出的基因突变50.6%(39/77)为TCIRG1基因,其定位于11q13.2[9],突变的类型主要包括错义/无义、小片段的插入/缺失、剪切/调控位点改变等[29]。TCIRG1基因编码液泡ATP酶(V-ATPase)a3亚基[30]。V-ATPase质子泵由介导ATP水解的细胞质水解结构域(V1)和促进细胞外酸化的跨膜质子易位结构域(V0)两个主要区域组成[2,31],在破骨细胞的皱褶缘表面高度表达[32],主要功能是将氢离子泵出胞外以酸化细胞外环境,实现离子跨膜转运,使钙、磷从骨组织中释放出来并沉着于类骨组织,形成新骨[33],此过程对骨骼的重塑起重要作用[1]。当TCIRG1基因发生突变时,V-ATPase蛋白不能发挥正常功能,破骨细胞无法吸收旧骨,软骨细胞钙化堆积,导致骨质变密、硬脆,骨髓腔缩小、消失。临床出现骨髓造血功能低下,引起肝、脾、淋巴结等髓外造血器官增大,长期贫血造成发育迟缓,体内钙质无法正常代谢而出现佝偻病。因骨质硬化,骨小梁结构不良,易发生骨折和骨髓炎;颅骨硬化可出现方颅、脑积水等表现;颅神经受压可导致视力障碍、眼球震颤、听力障碍、面瘫等。本文患儿父亲和母亲均携带杂合突变,分别为c.1360C>T(p.R454X)和c.722delC(p.T241fs)。结合患儿临床特点,明确患儿为复合杂合突变导致的IMO。
Qin等[34]报道,V-ATP酶复合物的不同亚基的缺失影响破骨细胞功能,导致IMO。Lucia等[35]研究发现所有具有TCIRG1突变的常染色体隐性遗传石骨症(ARO)患者都表现出严重的表型,可能由于大多数TCIRG1突变完全破坏蛋白质的功能。但是检测到少数仅有1个位点TCIRG1突变的患者与携带2个位点突变的患者有同样严重的表型,提示可能存在其他未检测到的突变。Sobacchi等[36]报道了1例临床诊断为ARO的8岁女孩,影像学表现符合石骨症,但没有出现神经或血液学缺陷。基因检测示TCIRG1基因纯合突变(c.1941+5G>A),导致产生多种异常转录物,但更重要的是,导致有限量的正常转录物,且这些低水平的正常TCIRG1蛋白可以减轻TCIRG1依赖性恶性ARO的临床表现。这个结果揭示了温和形式的TCIRG1-依赖性ARO的存在,为基因治疗提供了思路。TCIRG1基因的转录本是单独发挥作用还是被其他基因修饰?这种良性形式在接下来的几年里是否会进展,发生诸如神经压迫等并发症?TCIRG1基因型与临床表型的复杂相关性需要进一步研究。
CLCN7基因突变具有与TCIRG1基因突变致IMO相似的临床表现,但CLCN7突变的IMO患儿表现有更多的发育迟缓和惊厥表型[37]。Aker等[38]发现SNX10基因突变致IMO病例的视力障碍、贫血和骨质脆化较为常见。Ott等[39]研究发现OSTM1基因可引起非常严重的疾病表型,伴有频繁的中枢神经系统表现。RANK和RANKL属于肿瘤坏死因子配体超家族成员,其突变通过影响破骨细胞的分化使破骨细胞数目大幅度减少,导致IMO[40]。本文文献复习发现77例行基因检测的IMO患儿中,国内19例全部为TCIRG1基因突变,而国外各基因型均有报道,推测可能是不同种族的遗传背景及地域环境的差异所致。
IMO发病早、进展快、预后差,大部分患儿于6岁内死亡。造血干细胞移植(HSCT)是根治IMO的唯一方法,然而,由于HSCT无法改善已有的神经系统后遗症[41],尽早获得HLA基因型相合的供者是移植成功的关键。随着生殖医学的发展,采用体外受精并在胚胎移植前行遗传学诊断已成为避免家族性遗传病传至下一代的有效手段。本研究对1例IMO患儿进行突变筛查,检测到TCIRG1基因2个突变,丰富了石骨症的致病突变谱,为产前诊断学基因芯片提供了新的数据。
