两个同构的过渡金属锗磷酸盐化合物的合成与表征
2018-04-11陈长概黄春作
陈长概,黄春作
(厦门大学材料学院,福建 厦门 361005)
铝硅酸盐沸石在选择性催化和气体分离方面有重要的商业用途,具有类似结构的金属磷酸盐化合物也逐渐引起了人们的关注[1-3].与传统的铝硅酸盐分子筛相比,金属磷酸盐化合物更容易形成具有超大孔道的架状结构[4],且过渡金属元素价态可变,具有很好的氧化还原性能以及丰富的配位形式[4-6].此外,金属磷酸盐化合物也有着非常广泛的应用,如:具有36-元环超大孔道的天然矿物黄磷铁矿,被广泛运用于催化、吸脱附等领域[7];铝钴磷酸盐CoAPO-5和CoAPO-11可用作环己烷和对甲基苯酚自动氧化的催化剂[8-9];LiCoPO4和LiFePO4被广泛运用于锂电池正极材料等[10-11].
与其他金属磷酸盐相比,锗磷酸盐的研究仍然处于初级阶段.目前,已报道的锗磷酸盐化合物仅有60余例,且大多数锗磷酸盐为三维架状结构.然而,在这些数量有限的锗磷酸盐化合物中却发现了诸多优异的物理性能[10,12-15],如:具有沸石分子筛结构基元的锌锗磷酸盐(DABCO)·ZnGe(HPO4)3被用于气体分离和选择性催化[14];具有Nasicon型结构的化合物MⅠGe(PO4)3(MⅠ=Li,Na,K,Ag)被广泛运用于快离子导体材料[12,15]等.
基于过渡金属磷酸盐和锗磷酸盐的特点,倘若能利用过渡金属丰富的配位方式,将过渡金属与锗磷酸两者结合起来,有可能得到新的过渡金属锗磷酸盐化合物,这将有助于丰富锗磷酸盐体系,并可能具备潜在的物理应用.本研究通过水热法,成功合成了两个具有二维层状结构的过渡金属锗磷酸盐Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co,Fe),并采用粉末X射线衍射(powder X-ray diffraction,PXRD)、单晶X射线衍射、红外光谱分析和热分析方法对样品的结构、官能团和热稳定性进行表征.
1 实验部分
1.1 试 剂
氯化铯(CsCl)、六水合氯化钴(CoCl2·6H2O)、铁粉(Fe)、氧化锗(GeO2)、三正丙胺(TPA)、仲丁醇(2-BuOH)和85%(质量分数,下同)磷酸(H3PO4),均为分析纯,购于国药集团化学试剂有限公司和阿拉丁试剂(上海)有限公司.
1.2 合成方法
化合物1的合成步骤如下:首先将CsCl(0.169 g,1 mmol)、CoCl2·6H2O(0.119 g,0.5 mmol)和GeO2(0.104 g,1 mmol)溶解在装有0.25 mL水的聚四氟乙烯内衬(容量为15 mL)中,然后逐滴加入TPA(0.25 mL,1.8 mmol)和2-BuOH(4 mL,42 mmol),最后逐滴加入85%H3PO4(0.25 mL,3.6 mmol);同时将磁转子置于其中,利用磁力搅拌器搅拌加速溶解;溶液中n(CsCl)∶n(CoCl2·6H2O)∶n(GeO2)∶n(H3PO4)=1∶0.5∶1∶3.6,初始pH值约为5;搅拌30 min后,将聚四氟乙烯内衬放入合金钢套中,置于240 ℃烘箱中反应5 d;反应结束后,将其置于空气中自然冷却.将产物过滤后干燥,得到紫色块状晶体.
化合物2的合成步骤如下:首先将CsCl(0.169 g,1 mmol)、Fe粉(0.039 g,0.7 mmol)和GeO2(0.104 g,1 mmol)溶解在装有2 mL水的聚四氟乙烯内衬(容量为15 mL)中,然后逐滴加入TPA(2 mL,14.4 mmol)和2-BuOH(4 mL,42 mmol),最后逐滴加入85%H3PO4(0.75 mL,10.8 mmol);同时将磁转子置于其中,利用磁力搅拌器搅拌加速溶解;溶液中n(CsCl)∶n(Fe)∶n(GeO2)∶n(H3PO4)=1∶0.7∶1∶10.8,初始pH值约为5;搅拌30 min后,将聚四氟乙烯内衬放入合金钢套中,置于240 ℃烘箱中反应5 d;反应结束后,将其置于空气中自然冷却.将产物过滤后干燥,得到无色透明块状晶体.
在合成过程中,曾尝试将Fe或Co替换成Ni、Cu等过渡金属,但按照上述条件,始终无法得到与化合物1和2同构的晶体.同时,在合成过程中需要严格控制碱金属与过渡金属的量,只有当两者处在上述比例,才能够生成纯相目标化合物,否则容易生成其他杂质相.
1.3 样品表征
1) PXRD使用Bruker D8 Advance粉末衍射仪,工作电压40 kV,工作电流30 mA,采用Cu Kα射线作为光源,λ=0.154 06 nm,石墨单色器,测试范围为5°~60°.
2) 单晶X-射线衍射使用Bruker Smart CCD面探测衍射仪收集数据,采用Mo Kα射线作为光源,λ=0.071 073 nm,石墨单色器,在173 K下收集θ角度范围为2.68°~28.13°的数据.晶体结构由SHELXS-97程序[17-18],采用直接法和全矩阵最小二乘法解析.
3) 红外光谱分析采用KBr压片法,制样时将样品与KBr按质量比1∶100均匀混合并磨细,在干燥环境下使用Thermo Scientific Nicolet iS10傅里叶变换-红外(FT-IR)光谱仪进行测试,扫描范围为4 000~400 cm-1,配置ATR(attenuated total reflectance)配件,谱图分辨率为4 cm-1,扫描次数为32.
4) 热分析使用NETZSCH公司的TG-209F1热分析仪,测试的温度范围为35~900 ℃,升温速率为10 ℃/min,N2气氛下,N2流速为100 mL/min.
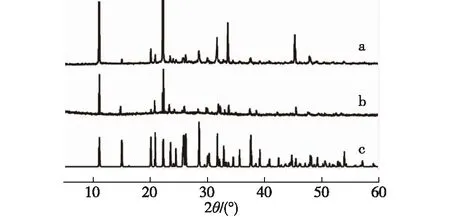
图1 化合物1(a)和化合物2(b)的实测PXRD谱图,及Cs[FeGe(OH)2 (H0.5PO4)2]的理论计算谱图(c)Fig.1 Measured PXRD spectra of compounds 1 (a) and 2(b), and theoretical calculation PXRD spectrum of Cs[FeGe(OH)2(H0.5PO4)2](c)
2 结果与讨论
2.1 晶体结构分析
从PXRD谱图可以发现化合物1(图1-a)和化合物2(图1-b)的衍射峰位置几乎完全相同,说明这两个化合物同构,因此这里仅以化合物1为例进行晶体结构分析.在光学显微镜下选出合适的晶体,使用BrukerSmart CCD面探测衍射仪收集衍射强度数据,得到衍射点1 854个,其中独立的衍射点为971个,符合I>2σ(I)条件的衍射点为951个.对数据进行LP因子和经验吸收校正,结构中的所有原子坐标均借助SHELX-97程序[17-18]采用直接法获得,原子坐标和占位度采用全矩阵最小二乘法精修至原子坐标的偏差因子R1=0.033 1,wR2=0.078 0,占位度S=1.074.通过单晶解析得到化合物1的化学式为Cs[CoGe(OH)2(H0.5PO4)2],化合物2的化学式为Cs[FeGe(OH)2(H0.5PO4)2],其中,化合物1的晶体学数据及精修参数列于表1.从PXRD谱图可以看出通过晶体解析得到的理论谱图(图1-c)与实测谱图(图1-a和1-b)匹配程度高.
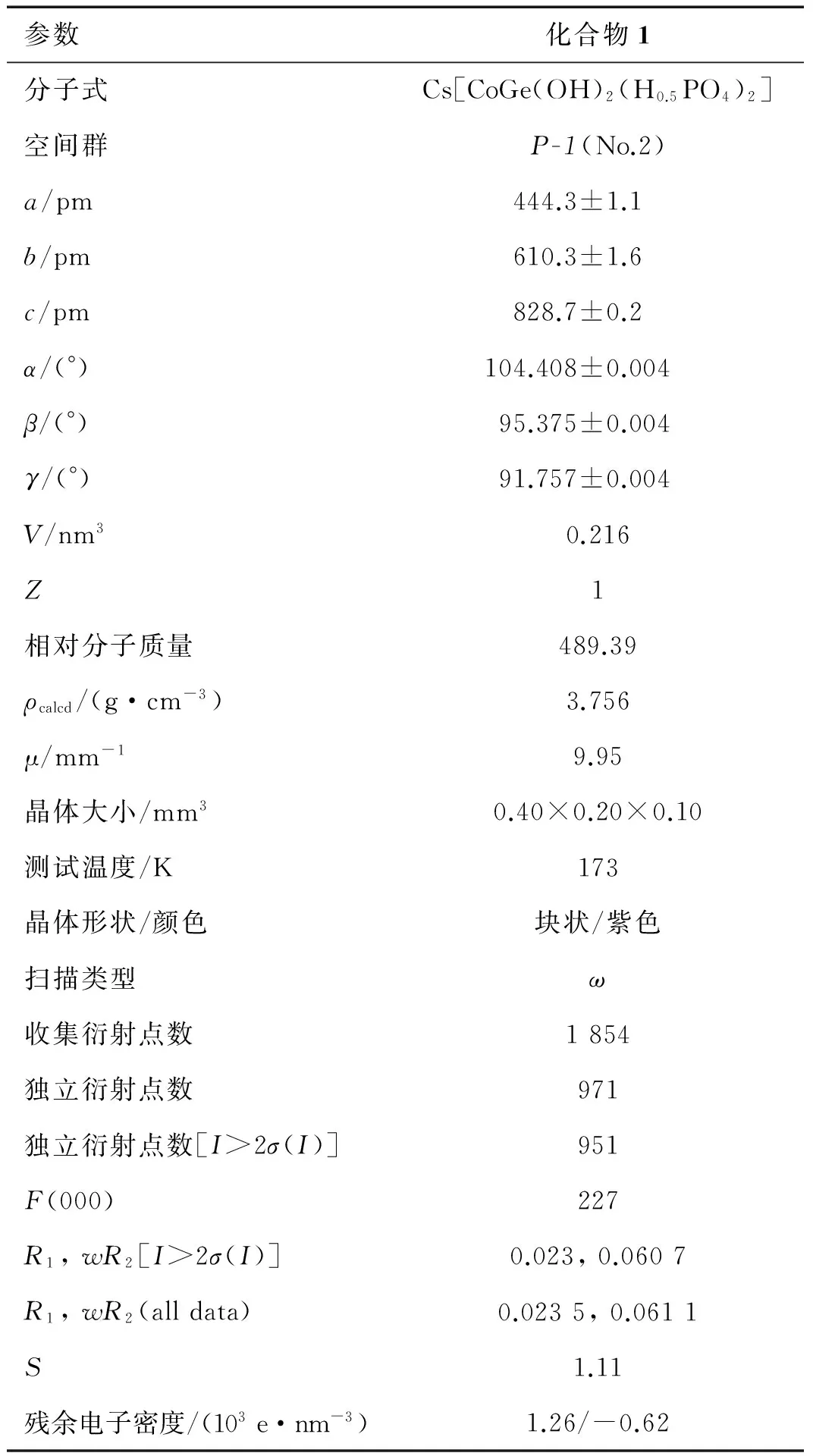
表1 化合物1的晶体学数据
2.2 晶体结构描述
两个化合物同构,因此以Cs[CoGe(OH)2(H0.5PO4)2]为代表进行晶体结构描述.在Cs[CoGe(OH)2(H0.5PO4)2]结构中,GeO4(OH)2八面体和CoO4(OH)2八面体通过共棱连接,在b轴方向形成一维链状结构(图2(a)).相邻的一维链状结构通过HPO4共角顶连接,在ab平面内形成一个二维层状结构,每个GeO4(OH)2八面体和CoO4(OH)2八面体的4个角顶通过共用O原子与4个HPO4四面体相连接(图2(b)).二维层状结构沿着c轴方向平行排列,层与层之间的Cs+不仅作为抗衡离子平衡层状结构的电荷,同时也与10个O原子相连最终形成一个伪三维结构(图2(c)).需要特别指出的是,通过单晶结构解析,发现H2原子占位度为0.5,H2原子与相邻层的O2原子相连形成氢键,此时H2原子平均位置处在对称氢键中,O—H键长为122.0 pm,比普通O—H键长大很多.但是正常情况下,由于H原子序数太低,X射线无法完全确定其位置,因此人为规定O—H键长为82 pm,类似的处理情况也曾在Li2Fe[(PO4)(HPO4)]晶体[19]中出现过.
Cs[CoGe(OH)2(H0.5PO4)2]的非对称结构单元如图3所示,包括1个Ge原子,1个Co原子以及1个P原子.其中Ge与4个O原子和2个羟基相连,形成GeO4(OH)2八面体,Ge—O键长为184.6~190.6 pm,平均键长为188.0 pm;Co原子与4个O原子和2个羟基相连,形成CoO4(OH)2八面体,Co—O键长为207.6~212.4 pm,平均键长为209.5 pm;P原子与3个O原子和1个羟基相连,形成变形的HPO4四面体,P—O键长为150.4~157.2 pm,平均键长为153.9 pm.上述Co—O、Ge—O、P—O键长与文献[20]报道的相符,处在合理范围内.根据BVS(bond valence sum)理论[21],Cs[CoGe(OH)2(H0.5PO4)2]中Cs、Co、Ge、P的价态分别为+1,+2,+4,+5价,具体的计算结果如表2所示.

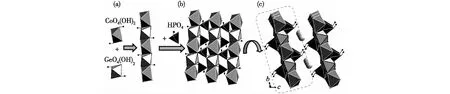
图2 Cs[CoGe(OH)2(H0.5PO4)2]的晶体结构Fig.2 Crystal structure of Cs[CoGe(OH)2(H0.5PO4)2]
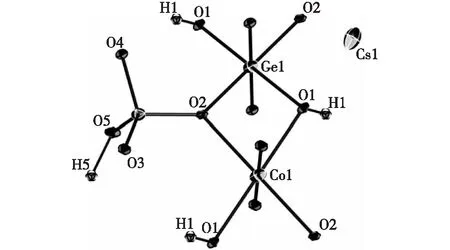
图3 Cs[CoGe(OH)2(H0.5PO4)2]的非对称结构单元中Co、Ge、P的配位环境Fig.3 Coordination environments of the cobalt,germanium and phosphorus atoms in the asymmetric unit of Cs[CoGe(OH)2(H0.5PO4)2]
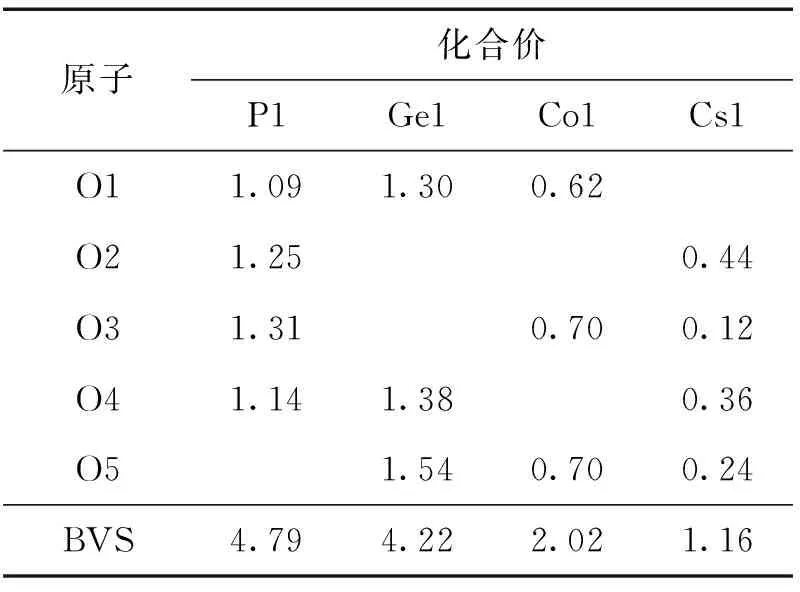
原子化合价P1Ge1Co1Cs1O11.091.300.62O21.250.44O31.310.700.12O41.141.380.36O51.540.700.24BVS4.794.222.021.16
2.3 红外光谱分析

(b) K2Co3(P2O7)2·2H2O;(c)Cs[CoGe(OH)2(H0.5PO4)2].图4 3种化合物的金属链比较Fig.4 The comparison of three kinds of metal chains
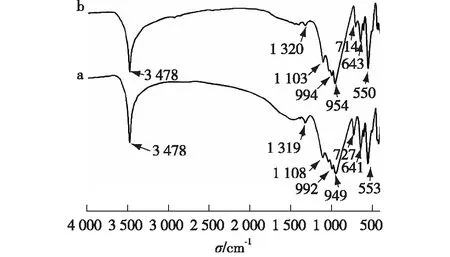
图5 化合物Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co(a),Fe(b))的FT-IR谱图Fig.5 FT-IR spectra of Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co(a),Fe(b))
Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co,Fe)的FT-IR谱图如图5所示.化合物Cs[CoGe(OH)2(H0.5PO4)2]中,3 478 cm-1处为O—H的非对称伸缩振动吸收峰;1 319 cm-1处为P—O—H的振动吸收峰;1 108 cm-1处为PO43-基团的非对称伸缩振动吸收峰;992和949 cm-1处为PO43-基团的对称伸缩振动吸收峰;727和553 cm-1处为PO43-基团的非对称弯曲振动吸收峰,这两处峰也可以归结于Ge—O的振动;641 cm-1处则为Ge—O的伸缩振动吸收峰.
Cs[FeGe(OH)2(H0.5PO4)2]中,3 478 cm-1处为O—H的非对称伸缩振动吸收峰;1 320 cm-1处为P—O—H的振动吸收峰;1 103 cm-1处为PO43-基团的非对称伸缩振动吸收峰;994和954 cm-1处为PO43-基团的对称伸缩振动吸收峰;714和550 cm-1处为PO43-基团的非对称弯曲振动吸收峰,这两处峰也可以归结于Ge—O的振动;643 cm-1处则为Ge—O的伸缩振动吸收峰.
2.4 热分析
图6为Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co,Fe)化合物在氮气中以10 ℃/min的速率从35 ℃加热至900 ℃的热重(TG)曲线.从图中可以看出,在350 ℃之前化合物的质量几乎不变,证明化合物中不含有H2O.在350~700 ℃范围内,两个化合物均存在1个失重台阶:其中Cs[CoGe(OH)2(H0.5PO4)2]的总失重为6.0%,与计算的每分子失去1.5个分子水的失重(5.52%)相当;而Cs[FeGe(OH)2(H0.5PO4)2]的总失重为5.9%,与计算的每分子失去1.5个分子水的失重(5.56%)相当.因此Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co,Fe)分子中只存在羟基.
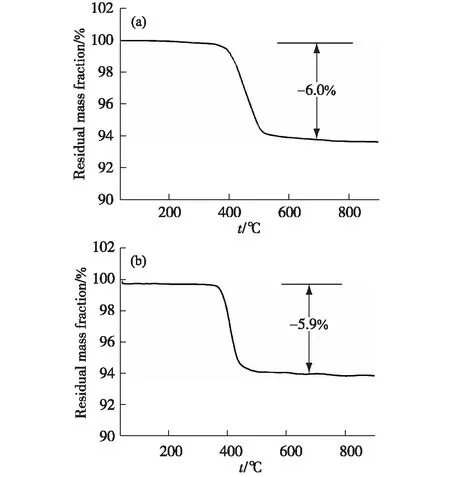
图6 Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co(a),Fe(b))在氮气中的TG曲线Fig.6 TG curves in nitrogen of Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co(a),Fe(b))
3 结 论
本研究利用水热法成功合成了两个新的过渡金属锗磷酸盐Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Fe,Co)化合物,对其进行了PXRD分析发现这两个化合物同构.以Cs[CoGe(OH)2(H0.5PO4)2]为代表,利用单晶X-射线衍射分析测定其结构并进行详细的结构描述,并对两个化合物进行了FT-IR和热分析,进一步确定了其结构特征.在两个化合物的结构中观察到由GeO4(OH)2八面体和MⅡO4(OH)2(MⅡ=Co,Fe)八面体共棱相连形成的一维链,但是由于Ge元素是主族金属,其d层电子数排满,姜-泰勒效应不明显,使得Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co,Fe)中的一维金属链相对较直.同时Cs[MⅡGe(OH)2(H0.5PO4)2](MⅡ=Co,Fe)是为数不多的具有二维层状结构的过渡金属锗磷酸盐.
参考文献:
[1]OLSON D H,HAAG W O,LAGO R M.Chemical and physical properties of the ZSM-5 substitional series[J].Journal of Catalysis,1980,61(2):390-396.
[2]CUNDY C S,COX P A.The hydrothermal synthesis of zeolites:history and development from the earliest days to the present time[J].Chemical Reviews,2003,103(3):663-701.
[3]OLSON D H,KOKOTAILO G T,LAWTON S L,et al.Crystal structure and structural related properties of ZSM-5[J].Journal of Physical Chemistry,1981,85(15):2238-2243.
[4]WILSON S T,LOK B M,MESSINA C A,et al.Aluminophosphate molecular sieves:a new class of microporous crystalline inorganic solids[J].Journal of the American Chemical Society,1982,104(4):1146-1147.
[5]CORMA A.From microporous to mesoporous molecular sieve materials and their use in catalysis[J].Chemical Reviews,1997,97(6):2373-2419.
[6]MASPOCH D,RUIZ M D,VECIANA J.Old materials with new tricks:multifunctional open-framework materials[J].Chemical Society Reviews,2007,36(5):770-818.
[7]MOORE P B,SHEN J.An X-ray structural study of cacoxenite,a mineral phosphate[J].Nature,1983,306:356-358.
[8]DAI P S E,PETTY R H,INGRAM C W,et al.Metal substituted aluminophosphate molecular sieves as phenol hydroxylation catalysts[J].Applied Catalysis A:General,1996;143(1):101-110.
[9]LIN S S,WENG H S.Liquid-phase oxidation of cyclo-hexane using CoAPO-5 as the catalyst[J].Applied Cataly-sis A:General,1993,105(2):289-308.
[10]AMINE K,YASUDA H,YAMACHI M.Olivine LiCoPO4as 4.8 V electrode material for lithium batteries[J].Electrochemical and Solid State Letters,2000,3(4):178-179.
[11]YAMADA A,CHUNG S C,HINOKUMA K.Optimized LiFePO4for lithium battery cathodes[J].Journal of the Electrochemical Society,2001,148(3):A224-A229.
[12]FENG J K,XIA H,LAI M O,et al.NASICON-structured LiGe2(PO4)3with improved cyclability for high performance lithium batteries[J].The Journal of Physical Chemistry C,2009,113(47):20514-20520.
[13]KOSEVA I,NIKOLOV V,PESHEV P.Effect of germanium doping on the morphology of flux grown Nb:KTiOPO4single crystals[J].Journal of Alloys and Compounds,2003,353(1/2):L1-L4.
[14]LI J M,KE Y X,ZHANG Y G,et al.((DABCO)·ZnGe(HPO4)3:the first zinco-germanophosphate with a unique asymmetric cage[J].Journal of the American Chemical Society,2000,122(25):6110-6111.
[15]TEREBILENKO K V,SLOBODYANIK N S,OGO-RODNYK I V,et al.Crystallization of MIGe2(PO4)3(MI=Na,K,Ag) from molten phosphate media[J].Crystal Research and Technology,2014,49(4):227-231.
[16]HUANG Y X,LIU B,WEN L,et al.Structural assembly from phosphate to germanophosphate by applying germanate as a binder[J].Inorganic Chemistry,2013,52:9169-9171.
[17]SHELDRICK G M.A short history of SHELX[J].Acta Crystallographica Section A,2008,64:112-122.
[18]SHELDRICK G M.Crystal structure refinement with SHELXL[J].Acta Crystallographica Section C:Structural Chemistry,2015,71:3-8.
[19]MI J X,BORRMANN H,ZHANG H,et al.Synthesis,magnetism,and crystal structure of Li2Fe[(PO4)(HPO4)] and its hydrogen position refinement[J].Zeitschrift fur Anorganische und Allgemeine Chemie,2004,630:1632-1636.
[20]HUANG Y X,ZHANG X,HUANG X,et al.Two isotypic transition metal germanophosphates M4Ⅱ(H2O)4Ge(OH)2(HPO4)2(PO4)2(MⅡ=Fe,Co):synthesis,structure,Mossbauer spectroscopy,and magnetic properties[J].Inorganic Chemistry,2012,51(5):3316-3323.
[21]BROWN I D,ALTERMATT D.Bond-valence parameters obtained from a systematic analysis of the inorganic crystal structure database[J].Acta Crystallographica Section B:Structural Science,1985,41:244-247.
[22]LIGHTFOOT P,CHEETHAM A K,SLEIGHT A W.Hydrothermal synthesis and crystal structure of a new layered phosphate,K2CO3(P2O7)2·2H2O[J].Journal of Solid State Chemistry,1990,85(2):275-282.
