高效液相色谱法同时测定水源水中三种微囊藻毒素
2018-04-11肖潺潺方向梅凡彪陈茂剑曾洁吴玲红利基林黄天壬黎远冬邓伟
肖潺潺 方向 梅凡彪 陈茂剑 曾洁 吴玲红 利基林,3 黄天壬,3 黎远冬,3 邓伟,3
人类生产和生活产生的大量氮、磷能引起水体富营养化,当富营养化程度严重的水体遇到适合藻类生长的温度和光照等条件时,会导致蓝藻水华频繁发生。而毒蓝藻水华的爆发会分泌一种次级代谢产物——藻毒素,可抑制其他物种在水体中生长,以确保藻类本身的生长优势[1]。微囊藻毒素(microcystins,MCs)为单环七肽毒素,由90多个异构体组成[2],具有强致癌性,若长期饮用含MCs的水,可对人体造成伤害,甚至引起肝癌发生[3]。水体中最常见的MCs为MC-LR、MC-RR和MC-YR,其中MC-LR毒性大于MC-RR和MC-YR[4]。MCs化学性质稳定,自然环境下降解非常缓慢[5]。目前自来水厂传统的处理工艺如沉淀、混凝、过滤、加氯并不能有效将其去除,饮用自来水的人群也有摄入微囊藻毒素危险[6]。酶联免疫吸附试验(enzyme linked immunosorbent assay,ELISA)的抗体与毒素存在交叉反应,可出现假阳性结果,对毒素类型鉴别不佳[7-8]。高效液相色谱(highperformance liquid chromatography,HPLC)具有良好的分离能力,可对多种微囊藻毒素进行精确地定性和定量测定[7]。但一次分离检测多种毒素的方法报道较少,且实验条件差别较大。本研究在以往研究的基础上,优化提取MCLR、MC-RR、MC-YR 3种MCs并采用HPLC同时检测,以进一步改进和完善水样中MCs的HPLC检测技术。
1 材料与方法
1.1 主要仪器和试剂
LC-20A高效液相色谱仪购自日本岛津有限公司,包括LC-20AB高压泵、SPD-20A紫外线检测器、CTO-20A柱温箱,恒温水浴锅购自北京西城新瑞仪器厂,真空泵、SUPELCO(500mg/6mL)购自北京康林有限公司,0.45μm有机滤膜购自天津津腾实验设备有限公司。微囊藻毒素-RR、LR标准品(纯度≥97%)购自北京索莱宝科技有限公司,微囊藻毒素-YR标准品(纯度≥98%)购自Alexis有限公司。甲醇和乙腈(色谱纯)、三氟乙酸(tallow fatty acid,TFA)(分析纯)购自南宁科利尔生物有限公司。
1.2 标准样品配制
MC-RR、MC-LR标准储备液:将50μg纯MC-RR、MC-LR标准物质用0.5mL甲醇溶解配制成100μg/mL标准储备液,MC-YR储备液10μg/mL,可在1~4℃保存6个月,将MC-RR、MC-LR、MC-YR3种标准品逐级稀释配制成0.05μg/mL、0.125μg/mL、0.25μg/mL、0.5μg/mL、1.0μg/mL系列浓度标准溶液,在色谱分析条件下依次测定。
1.3 环境水样采集与前处理
水样采用横式采水器采集水面下0.5 m处表层水样,经砂芯漏斗去除大颗粒杂质,置于4℃避光保存。量取500 mL水样经0.45μm滤膜减压过滤。胞内MCs提取步骤:滤膜在-80℃和37℃反复冻融3次,待测。胞外MCs固相萃取步骤:依次用10mL 100%甲醇、10mL超纯水活化固相萃取柱;再将过滤后的水样流经固相萃取柱进行富集浓缩,流速为4.0mL/min;依次用10mL超纯水和20mL 20%甲醇溶液淋洗固相萃取柱;以15 mL 80%甲醇溶液(加入0.05%TFA)将固相萃取柱上的MCs洗脱收集。15mL收集液在普通恒温水浴锅中60℃水浴,同时缓慢吹入氮气吹脱,蒸发浓缩吹干,-20℃冷冻避光保存,待分析。分析前溶于1mL超纯水。
1.4 淋洗液、洗脱液、流动相和流动相磷酸的含量选择
对于淋洗液浓度的选择,用10 mL超纯水淋洗经活化处理和上样浓缩后的小柱,分别采用10%、15%、20%、30%甲醇溶液20mL淋洗固相萃取柱,收集不同浓度的淋洗液,HPLC检测比较其中的毒素和杂质。对于洗脱液的浓度,本实验在其他条件不变的情况下对比了70%、80%、90%浓度甲醇洗脱液。选择以下4种流动相进行比较实验:⑴0.1%磷酸水溶液-乙腈(50∶50);⑵0.1%磷酸水溶液-乙腈(70∶30);⑶0.1%磷酸水溶液-乙腈(67∶33);⑷0.1%磷酸水溶液-乙腈(65:35)。对以下3种磷酸含量的流动相进行比较实验:⑴:水溶液-乙腈(67∶33);⑵:0.1%磷酸水溶液-乙腈(67∶33);⑶:0.2%磷酸水溶液-乙腈(67∶33)。采用梯度洗脱方式,对比3种流动相在保留时间、分离度、响应强度方面的差异。
1.5 HPLC测定条件
色谱柱:Inersustain ODS(4.6mm×250.0mm,5μm),在238 nm波长紫外线检测器检测,柱温45℃。流动相:0.1%磷酸水溶液-乙腈(67∶33),流速1.0 mL/min,梯度洗脱,进样量20μL。
1.6 统计学方法
采用SPSS 20.0统计分析软件对数据进行处理。分别以MC-RR、MC-LR、MC-YR 3种标准样品的色谱峰面积为因变量,标准样品浓度为自变量进行简单线性回归分析并计算相关系数。
2 结果
2.1 样品预处理的优化
2.1.1 淋洗液的优化本实验采用10%、15%、20%、30%甲醇溶液20mL淋洗固相萃取柱,收集不同浓度的淋洗液,用HPLC检测比较其中的毒素和杂质。经比较,20%甲醇淋洗液较合适。
2.1.2 洗脱液的优化本实验在其他条件不变的情况下对比70%、80%、90%浓度甲醇洗脱液,结果显示,80%甲醇溶液为洗脱液的方案最佳。
进一步对洗脱步骤中的一些操作细节进行优化,结果发现连续洗脱较分批次洗脱效果更佳,但应尽量避免小柱在洗脱过程中吹干。同时,洗脱液中加入TFA对回收率有一定影响,80%甲醇溶液洗脱液中加入0.05%TFA能明显提高毒素的回收率,特别是MCRR的回收率,而且重现性较好,在同一条件下平行测定6次不同洗脱液的加标回收率,结果见表1。

表1 不同洗脱液的平均加标回收率比较(n=6,%)
2.2 色谱条件的优化
2.2.1 流动相及流动方式的选择由于MCs易溶于水和乙腈,因此选择水-乙腈作为流动相进行实验。结果表明,选择0.1磷酸水溶液-乙腈(67∶33)流动相峰形好,响应值高,出峰时间快,在20min内可较好地分离微囊藻毒素,因此选择0.1磷酸水溶液-乙腈(67:33)。
2.2.2 流动相磷酸的含量对磷酸含量的流动相进行比较,结果表明,以水溶液-乙腈(67∶33)流动相分析MCs时保留时间较长,且MC-LR、MC-YR 2种毒素峰形和分离度均较差,加入磷酸后离子化效率明显增高,出峰时间快,响应强度高,且峰形好。本研究采用0.1磷酸水溶液-乙腈(67:33)流动相(pH值为3),可在20 min内分离MC-RR、MC-LR、MC-YR,保留时间分别为7.448 min、15.291 min、18.485 min,结果见图1。
2.2.3 标准曲线方程与检出限按照优化后的色谱条件,以色着峰面积对标准样品浓度求得线性回归方程以及相关系数(表2)和标准曲线(图2)。按信噪比(S/N)=3计算本方法MC-RR、MC-YR和MC-LR的检出限(limitof detection,LOD),分别为0.037μg/L、0.056 μg/L和0.028μg/L。
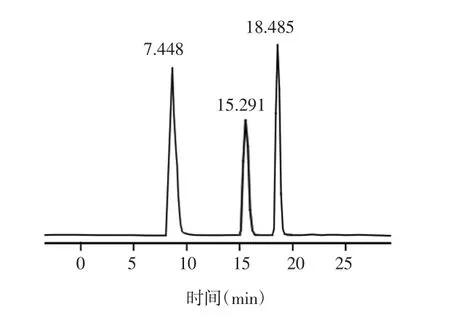
图1 MC-RR、MC-YR、MC-LR标准样品液相色谱图
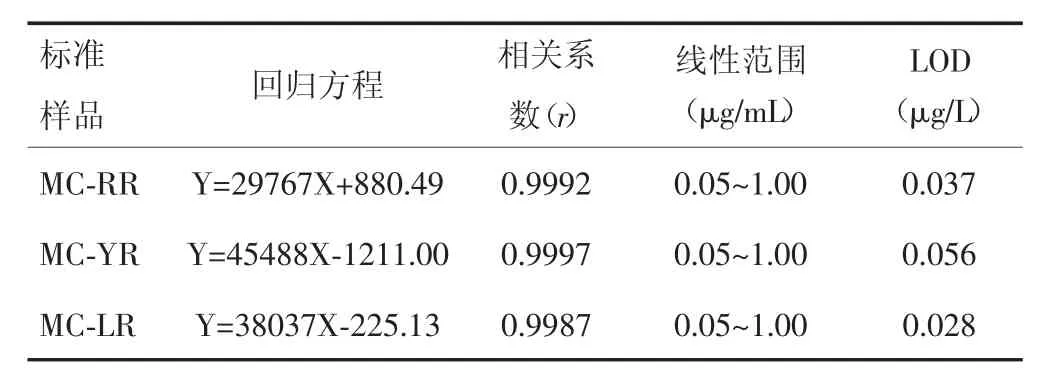
表2 MC-RR、MC-YR、MC-LR的回归方程、相关系数和检出限
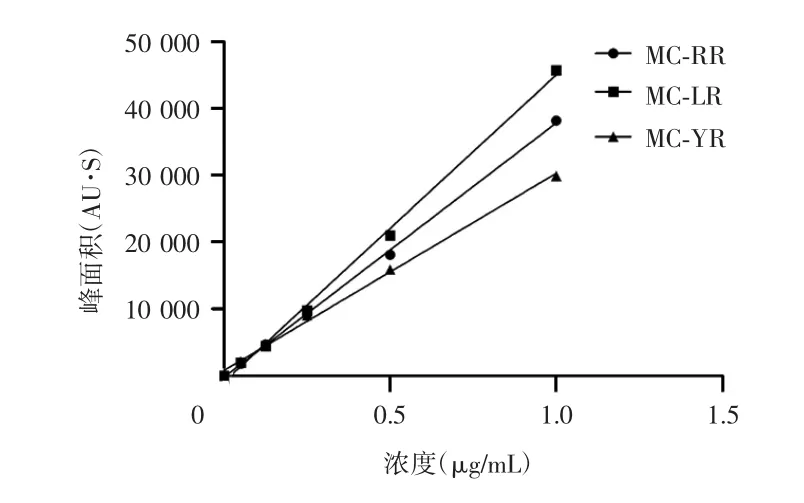
图2 HPLC检测MC-RR、MC-YR、MC-LR的标准曲线
2.2.4 回收率和精密度应用本方法检测某水源水、末梢水和纯水中的MCs,样本均加入0.1μg/L的MC-RR、MC-LR、MC-YR标准样品,经吸附、洗脱、浓缩处理后,每个样品在选定的色谱条件下平行测定6次,结果见表3。其中MC-RR测定结果的相对标准偏差(relative standard deviation,RSD)为5.0%~8.2%,平均回收率为80%~100%;MC-YR测定结果的RSD为6.2%~8.1%,平均回收率为80%~90%;MC-LR测定结果的RSD为5.4%~9.6%,平均回收率为93%~110%。回收率均在80%~120%之间,满足GB 5749-2006《生活饮用水标准检验方法水质分析质量控制》对测量准确度的要求。
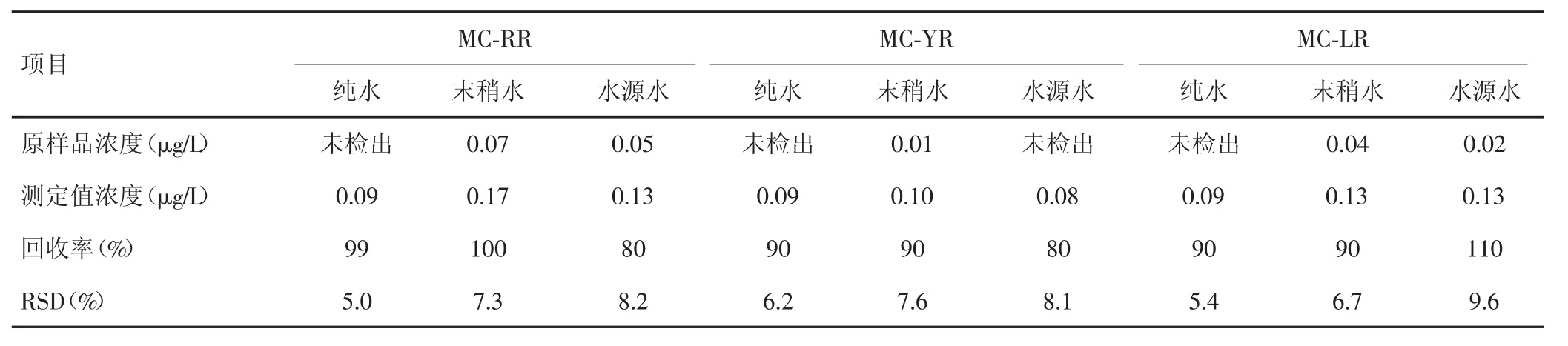
表3 HPLC检测MC-RR、MC-YR及MC-LR的精密度和回收率(n=6)
3 讨论
水体富营养化不断加剧可使有毒藻类大量繁殖,水体中藻毒素特别是MCs污染日益严重。由于传统的处理工艺如沉淀、混凝、过滤、加氯均不能有效将其去除,人们可通过直接接触、食物链传递和饮水3种途径长期低剂量接触,因此,探讨MCs对人类健康的危害尤为必要,而对MCs进行定性和定量是先决条件之一。MCs是细胞内毒素,在细胞内合成,只有细胞壁破裂后才释放出来。对其进行检测时,特定的细胞预处理至关重要。目前报道较多的破坏细胞壁的方法是反复冻融和超声破碎机。Ramanan等[9]研究发现,超声破碎虽可释放更多的毒素,但可引起多肽结构断裂而使其降解,MCs的量并不能明显增加。所以本实验在-80℃和37℃反复冻融3次,结果发现产毒总量高且省时。
对于淋洗液的选择,甲醇浓度越低的淋洗液洗脱能力越差,其中杂质和毒素含量亦越低[10]。如低于15%的甲醇淋洗液中只含少量色素等杂质,保留在小柱中的杂质势必造成后续检测中MCs峰被严重干扰。而浓度为30%的甲醇淋洗液中杂质含量最大,色谱图起始基线很高,但同时又有少量MC-RR、MC-YR被洗脱检出,导致后续检测中毒素回收率偏低。本研究发现20%的甲醇淋洗液较为合适。
对于洗脱液浓度的选择,本研究结果发现用浓度为90%的甲醇溶液洗脱,其洗脱收集液的色谱图上杂质较多,毒素峰被严重干扰。而70%和80%甲醇洗脱液洗脱效率较接近,70%甲醇洗脱液的回收率略高,但考虑到后续浓缩步骤70%甲醇溶液洗脱下毒素的收集液较难吹干,因为洗脱剂中水相比例增加会使蒸发浓缩过程时间增加几倍,延长实验周期,综合考虑省时、低干扰以及高回收等因素,发现以80%甲醇溶液作为洗脱液最佳。TFA可调节pH值,促进MCs多肽质子化,并与之形成离子对,削弱MCs多肽与硅胶表面硅醇基的相互作用力,使其更易被洗脱[10]。而且TFA的紫外背景吸收小,易挥发,也有利于减少分析干扰。所以本实验在80%甲醇溶液洗脱液中加入0.05%TFA,结果能明显提高毒素的回收率和减少杂质干扰。
理论上,HPLC谱可分辨出几十种肽的混合物,但实际上分析、分离微小差别异构体的混合物仍非常困难,因此在分离这一类化合物时通常使用酸性流动相以提高色谱效率。比如加入磷酸调节流动相,较低的pH值可使肽中的羧酸基质子化,增加电离效果[11]。有报道[12-13],随着水-乙腈流动相酸度增大,可明显改善MCs峰形,但MC-RR出峰过早会和实际样品中先出来的杂质混合,而且色谱柱也有一定的耐酸限度,因此要选择合适的酸度,既要有较好的峰形和分离度,也要对色谱柱加以保护。本实验采用0.1磷酸水溶液-乙腈(67∶33)流动相(pH值为3),可在20 min内分离MC-RR、MC-LR、MC-YR,保留时间分别为7.448min、15.291 min、18.485 min,在0.05~1.0μg/mL范围内线性良好,回收率均在80%~110%之间,RSD为5.0%~9.6%,LOD分别为0.37、0.028和0.056μg/mL,说明本法可行、有效。
本实验采用优化的HPLC方法同时测定水样中MC-LR、MC-RR和MC-YR,萃取时采用20%的甲醇溶液淋洗,80%甲醇溶液(加入0.05%TFA)洗脱,能明显提高MCs回收率,且重现性好。HPLC进样分析选用0.1%磷酸水溶液-乙腈(67:33)作为流动相进行梯度洗脱,对MC-LR、MC-RR和MC-YR分离效果较好,保留时间适中。本法解决了图谱杂质多,目标化合物的色谱峰分离不理想的问题,同时灵敏度高,精密度、准确度满足现行国内及国际水质检测标准要求,对微囊藻毒素HPLC检测技术的推广应用具有重要参考价值。
[1] 李科志,邓伟,李云西,等.广西肝癌高发区不同水源微囊藻毒素含量调查[J].中国癌症防治杂志,2016,8(6):387-389,390.
[2] Zegura B.An overview of themechanismsofmicrocystin-lrgenotoxicity and potential carcinogenicity[J].MiniRev Med Chem,2016,16(13):1042-1062.
[3] Saoudi A,Brient L,Boucetta S,et al.Management of toxic cyanobacteria for drinking water production of Ain Zada Dam[J].Environ Monit Assess,2017,189(7):361.
[4] Bi X,Dai W,Zhang S,et al.Effects of toxic Microcystis genotypes on natural colony formation and mechanism involved[J].Water Sci Technol,2017,76(3-4):885-894.
[5] 丁新良,何恩奇,钮伟民,等.太湖水体中微囊藻毒素-LR污染状况调查[J].现代预防医学,2012,39(17):4375-4377.
[6] Dziga D,Maksylewicz A,Maroszek M,et al.The biodegradation of microcystins in temperate freshwater bodies with previous cyanobacterial history[J].Ecotoxicol Environ Saf,2017,145:420-430.
[7] 孙睿华.微囊藻毒素的高效液相色谱测定法若干问题的探讨[J].环境与健康杂志,2015,32(5):448-449.
[8] Trifiro G,Barbaro E,Gambaro A,et al.Quantitative determination by screening ELISA and HPLC-MS/MS of microcystins LR,LY,LA,YR,RR,LF,LW,and nodularin in the water of Occhito lake and crops[J].Anal Bioanal Chem,2016,408(27):7699-7708.
[9] Ramanan S,Tang J,Velayudhan A.Isolation and preparative purification ofmicrocystin variants[J].JChromatogr A,2000,883(1-2):103-112.
[10] 龚黎明.高效液相色谱法检测微囊藻毒素的条件优化及应用研究[J].资源信息与工程,2017,32(2):178-179.
[11] Birungi G,Li SF.Determination of cyanobacterial cyclic peptide hepatotoxins in drinking water using CE[J].Electrophoresis,2009,30(15):2737-2742.
[12] 邹康兵,向彩红,董玉莲,等.超高效液相色谱-串联质谱法测定饮用水中微囊藻毒素[J].化学分析计量,2017,26(1):42-46.
[13] 李诗言,王扬,王鼎南,等.通过型固相萃取-液相色谱-串联质谱法同时快速测定鱼肉中7种微囊藻毒素[J].色谱,2017,35(8):794-800.
