硒化肿节风浸膏残渣多糖的制备、工艺优化及抗肿瘤活性研究
2018-03-20刘雅萌戴士杰高向东
胡 成,刘雅萌,戴士杰,刘 玮,高向东
(中国药科大学生命科学与技术学院 江苏省生物药物成药性研究重点实验室,南京 210009)
硒作为人体所必需的微量元素,广泛存在于人体的多种酶中,如谷胱甘肽过氧化物酶(GPx)、硫氧化还原蛋白还原酶(TrxR)和甲酸脱氢酶等[1],同时硒元素具有抗氧化、抗炎、化学预防、抗病毒和抗肿瘤等多种活性[2]。硒元素以无机硒和有机硒两种形式存在于自然界中[3]。有机硒相比于无机硒,更易于在动物的消化道中吸收,并在尿中排泄,从而有着更高的生物利用度。同时,有机硒与无机硒相比,其毒性也低得多。近年来,有机硒化合物由于其特殊的结构以及显著的生物活性,愈发受到人们的关注。因此,越来越多的有机硒化合物作为天然食物补充剂成为肿瘤治疗和预防过程中的重要膳食补充[4]。多篇报道称硒化多糖作为有机硒化合物,在抗肿瘤、免疫调节、降低血糖、降低血脂和抗菌等方面有着更强的活性[5]。然而,天然硒化多糖十分稀少,因此人们开始着眼于对多糖进行硒化修饰。
肿节风[Sarcandraglabra(Thunb.) Nakai]为金栗兰科(Chloranthaceae)植物草珊瑚的干燥全草[6],作为传统中药,用于治疗多种疾病[7]。调研发现,中国有30多家制药企业生产超过10种肿节风药物。在这些药物的制备过程中,乙醇提取工艺产生了上百吨乙醇沉淀的废弃残渣,对残渣的“三无”处理也消耗了大量的人力物力及财力。然而,实验室前期研究表明这些废弃残渣中存在着大量多糖,但由于残渣的溶解性差,使其中的多糖成分较难得到深加工利用。近年来,由于多糖经过化学修饰后,其结构发生了部分改变,其理化性质、生物活性随之改变,修饰后的多糖具有更好的溶解性从而可展示更多的药理活性,同时具有较低的毒性,因此对多糖进行化学修饰已经成为多糖在药学研究和应用的一种重要的改良方法。硒化修饰后产生的硒化多糖是由硒和多糖结合组成,有着双重药理作用,然而又非两种活性的简单相加,硒化多糖往往会比原多糖具有更好的活性。目前,各种新型有机硒多糖正在不断地被合成出来,现有硒多糖的药理作用也被逐步阐明,有望成为高效低毒的新型药物,利用硒多糖独特的化学和生物学性质来开发新的药物已引起越来越多的人们的高度关注。根据本课题组前期研究发现,肿节风浸膏残渣中含有大量多糖成分,对其进行硒化修饰将有可能提高其抗肿瘤活性,拓宽其利用范围[8]。因此,本研究以肿节风浸膏残渣粗多糖为原料对其进行硒化修饰,采用正交设计方法建立了硒化多糖的制备工艺,并对9种不同硒化度的硒化肿节风浸膏残渣多糖的纯度、相对分子质量、初步的结构特征及抗肿瘤活性进行了比对研究。
1 材 料
1.1 试 剂
肿节风浸膏残渣(江西江中集团提供);亚硒酸钠、MTT、氟尿嘧啶(美国Sigma公司);硒元素标准溶液(国家标准物质研究中心);胰酶(美国Biosharp公司);胎牛血清(浙江天杭生物科技有限公司);DMEM培养基(美国Gibco公司);其他试剂均为市售分析纯。
1.2 仪 器
752紫外可见分光光度计(上海菁华科技仪器有限公司);WX-8000微波消解系统(上海屹尧仪器科技发展有限公司);Nicolet 6700红外光谱仪、Multiskan全波长酶标仪、Thermo Scientific Sorvall ST 16离心机(美国Thermo fisher公司);AFS-9800原子荧光光谱仪(北京科创海光仪器公司);Varian 500核磁共振仪(美国Varian公司);BS214D电子天平(德国赛多利斯公司);Agilent 1260高效液相色谱仪(美国Agilent公司)。
1.3 细胞株
HT-29人结肠癌细胞株、HepG2人肝癌细胞株、L02人正常肝细胞株、HeLa人宫颈癌细胞株(中国科学院典型培养物保藏委员会细胞库)。
2 方 法
2.1 硒化肿节风浸膏残渣多糖的制备
2.1.1 肿节风浸膏残渣粗多糖(SERP)的制备 肿节风浸膏残渣经干燥后溶于蒸馏水中,制备成10 mg/mL的溶液,加入无水乙醇至醇浓度为30%,过夜醇沉后经4 000 r/min离心20 min,收集沉淀,真空干燥后得SERP。
2.1.2 硒化肿节风浸膏残渣多糖的制备 将SERP溶解于一定浓度HNO3溶液中,配置成10 mg/mL的溶液,加入适量BaCl2和亚硒酸钠2 g,在一定温度条件下搅拌反应适当时间。反应结束后加入Na2CO3调pH至7~8,滴入适量的Na2SO4后离心除去BaSO4。用蒸馏水透析72 h,直到透析外液中加入过量抗坏血酸不再生成橙色单质硒,未透过液经低温减压浓缩、 冷冻干燥后使用DEAE-32离子交换柱色谱纯化,得到硒化肿节风浸膏残渣多糖。
2.1.3 影响SERP硒化修饰的因素考察
反应时间 按照“2.1.2”项方法,在反应温度70 ℃、BaCl21.0 g、HNO3质量分数1.0%的条件下,考察反应时间分别为6、7、8、9、10 h时对SERP硒化修饰工艺的影响。
反应温度 按照“2.1.2”项方法,在反应时间8 h、BaCl21.0 g、HNO3质量分数为1.0%的条件下,考察反应温度分别为40、50、60、70、80 ℃时对SERP硒化修饰工艺的影响。
HNO3质量分数 按照“2.1.2”项方法,在反应温度70 ℃、反应时间8 h、BaCl21.0 g的条件下,考察HNO3质量分数分别为0.5%、1.0%、1.5%、2.0%、2.5%时对SERP硒化修饰工艺的影响。
BaCl2用量 按照“2.1.2”项方法,在反应温度70 ℃、反应时间8 h、HNO3质量分数1.0%的条件下,考察BaCl2用量分别为0、0.5、1.0、1.5、2.0 g时对SERP硒化修饰工艺的影响。
2.1.4 制备工艺优化 在单因素实验的基础上,采用正交实验法优化SERP硒化修饰工艺。以反应时间、反应温度、HNO3质量分数、BaCl2用量为考察因素进行四因素三水平正交实验,以校准后总糖含量、产率、对于HepG2人肝癌细胞的半数抑制率和硒元素含量为指标进行考察。
2.2 硒化肿节风浸膏残渣多糖的理化性质和结构表征
2.2.1 中性糖、酸性糖含量测定及总糖含量的校准 参照文献中硫酸-苯酚法测量样品中性糖含量[9],间羟联苯测量样品中糖醛酸的含量[10]。总糖含量的校准:根据样品单糖组成中中性糖与酸性糖的比例校正偏差,计算后得到校准后总糖含量。
2.2.2 硒元素含量测定 将硒化肿节风浸膏残渣多糖,置于聚四氟乙烯管中,加入浓HNO3和30% H2O2,在微波消解系统中消化,以SERP作为空白对照。消解结束后,用原子荧光光谱仪测定其硒元素含量[11]。
2.2.3 样品纯度及相对分子质量测定 将硒化肿节风浸膏残渣多糖通过高效液相色谱仪进行纯度及相对分子质量测定。流动相为H2O,流速1 mL/min,柱温为37 ℃,检测器温度为40 ℃。取不同相对分子质量的右旋糖酐标准品经0.45 μm微孔滤膜过滤后进样,通过GPC分析软件获得相对分子质量校准曲线。将样品以相同浓度进样,分析色谱图,获得样品纯度信息,并计算获得其相对分子质量。
2.2.4 紫外光谱分析 配制0.1 mg/mL SERP及其硒化产物的水溶液,以蒸馏水为空白对照,于200~700 nm范围内进行扫描测定。
2.2.5 红外光谱分析 SERP及其硒化产物经干燥后与KBr按1∶200质量比充分混合、研磨,采用溴化钾压片法在红外光谱仪上于4 000~400 cm-1区间扫描。
2.2.6 热重分析 采用综合热重分析仪对SERP及其硒化产物进行热分析,以10 ℃/min的速率升温,范围控制在30~300 ℃,气体为静态N2,参比物为Al2O3。
2.3 抗肿瘤活性分析
选择人肺癌细胞A549、人胃癌细胞BGC-823、人宫颈癌细胞HeLa,人结肠癌细胞HT-29,人肝癌细胞HepG2,人正常细胞L02进行抗肿瘤活性筛选,细胞均培养在含有10%胎牛血清及双抗的完全培养基中,环境温度37 ℃,湿度95%,5% CO2。通过MTT法检测不同培养时间(24,48 h)下SERP及其硒化产物对各个细胞株的体外生长抑制效果。
3 结 果
3.1 SERP的制备
从肿节风浸膏残渣30 g中提取获得SERP(7.68±0.49) g,样品呈棕黄色。
3.2 反应时间、温度以及HNO3、BaCl2用量对SERP硒化修饰的影响
反应时间对SERP硒化修饰产率的影响如图1-A所示。在反应温度70 ℃、BaCl21.0 g、HNO3质量分数1.0%的条件下,当反应时间为8 h时,硒化修饰产率最高,随后产率将会有所下降。引起该现象的原因可能为:反应时间太短导致反应不充分,硒与原糖不能有效结合;反应时间太长,多糖在高温、强酸条件下会产生降解,导致产率降低。
反应温度对SERP硒化修饰的影响如图1-B所示。当温度低于70 ℃时,随着反应温度的升高,硒化产物的产率逐渐增加,而高于70 ℃后,其产率锐减。这可能是因为多糖在高温条件下发生了降解,因此产率降低明显。
图1-C为反应温度70 ℃、反应时间8 h、BaCl21.0 g条件下,不同质量分数HNO3对SERP硒化修饰的影响。当HNO3质量分数低于1.0%时,硒化产物的产率随着HNO3质量分数的提高而升高,但当HNO3质量分数超过1.0%时,产率则在逐步下降。由于HNO3属于强酸,酸度过高易造成多糖的降解,使硒化反应难以发生,同时HNO3质量分数太低又不利于催化反应。
BaCl2用量对SERP硒化修饰的影响如图1-D所示。当固定反应温度为70 ℃、反应时间为8 h、HNO3质量分数为1.0%时,随着BaCl2加入量的提高,硒化产物的产率逐步升高,在BaCl2加入量为1.0 g时达到峰值。但随后随着BaCl2加入量的增加,其产率逐渐下降。这可能是因为BaCl2的加入对硒化反应起到了催化作用,但当BaCl2过量时,阻碍了硒与原糖的反应,从而导致产率降低。


3.3 硒化肿节风浸膏残渣多糖的制备工艺优化结果
经过四因素三水平正交实验,最终得到了9种硒化肿节风浸膏残渣多糖产物S1~S9,方差分析结果见表1,2。正交试验结果显示,影响硒化产率的因素由大到小依次为HNO3质量分数、反应温度、BaCl2用量、反应时间;影响产物中硒含量的因素由大到小依次为BaCl2用量、反应温度、反应时间、HNO3质量分数。但考虑其抗肿瘤活性后,在产率相似,硒含量适中的情况下,将活性较高,产率相差较小的条件拟选为最优方案,即反应温度为70 ℃、反应时间为7 h、HNO3质量分数为1.0%、BaCl2用量为1.5 g。在该优化反应条件下,重复3次验证实验,硒化肿节风浸膏残渣多糖的总糖含量达到(92.30±3.22)%,硒含量为(929.77±12.51) μg/g,产率为(22.15±0.66)%,对于HepG2的半数抑制率为(123.33±23.02) μg/mL。
Table1 Variance analysis (ANOVA) and significant test of orthogonal experiment result on yield

FactorSddfVarianceFSigTemperature19822299112031∗∗Time654323271670BaCl2920224601942∗HNO3260712130362671∗∗
*P<0.05;**P<0.01
Table2 ANOVA and significant test of orthogonal experiment result on selenium content

FactorSddfVarianceFSigTemperature2521249621260624810960∗∗Time14237109271185546189∗∗BaCl24285992822142996418632∗∗HNO37636622381831332
*P<0.05;**P<0.01
3.4 纯度及相对分子质量测定结果
硒化肿节风浸膏残渣多糖S1~S9的高效液相色谱图如图2所示,其重均相对分子质量(Mw)、数均相对分子质量(Mn)、Z均相对分子质量(Mz)和分散系数(PD)如表3所示。由图2和表3可以看出,9种硒化肿节风浸膏残渣多糖的纯度和相对分子质量基本一致。
3.5 紫外光谱分析
图3为SERP及其9种硒化产物的紫外吸收光谱图。如图所示,SERP在230 nm区域内无吸收峰,而硒化修饰后9种产物均在230 nm处产生吸收。该结果提示硒化修饰向原糖分子中引入了亚硒酸根,继而可能形成了硒化肿节风浸膏残渣多糖[12]。

Figure2 HPLC chromatograph of selenylated polysaccharides 1-9 (S1-S9)
Table3 Weight-average molecular weight(Mw),number-average molecular weight(Mn),Z-average molecular weight(Mz) and polydispersity (PD)of S1-S9

ContentMwMnMzPDS1134216312908891392106103972S2133743212870881386171103911S3133935212888901389358103915S4133806112877661386266103906S5134116612891111396242104038S6133894612885701387231103909S7134018112900051390257103890S8133841812872071390338103978S9133502412849381383568103898
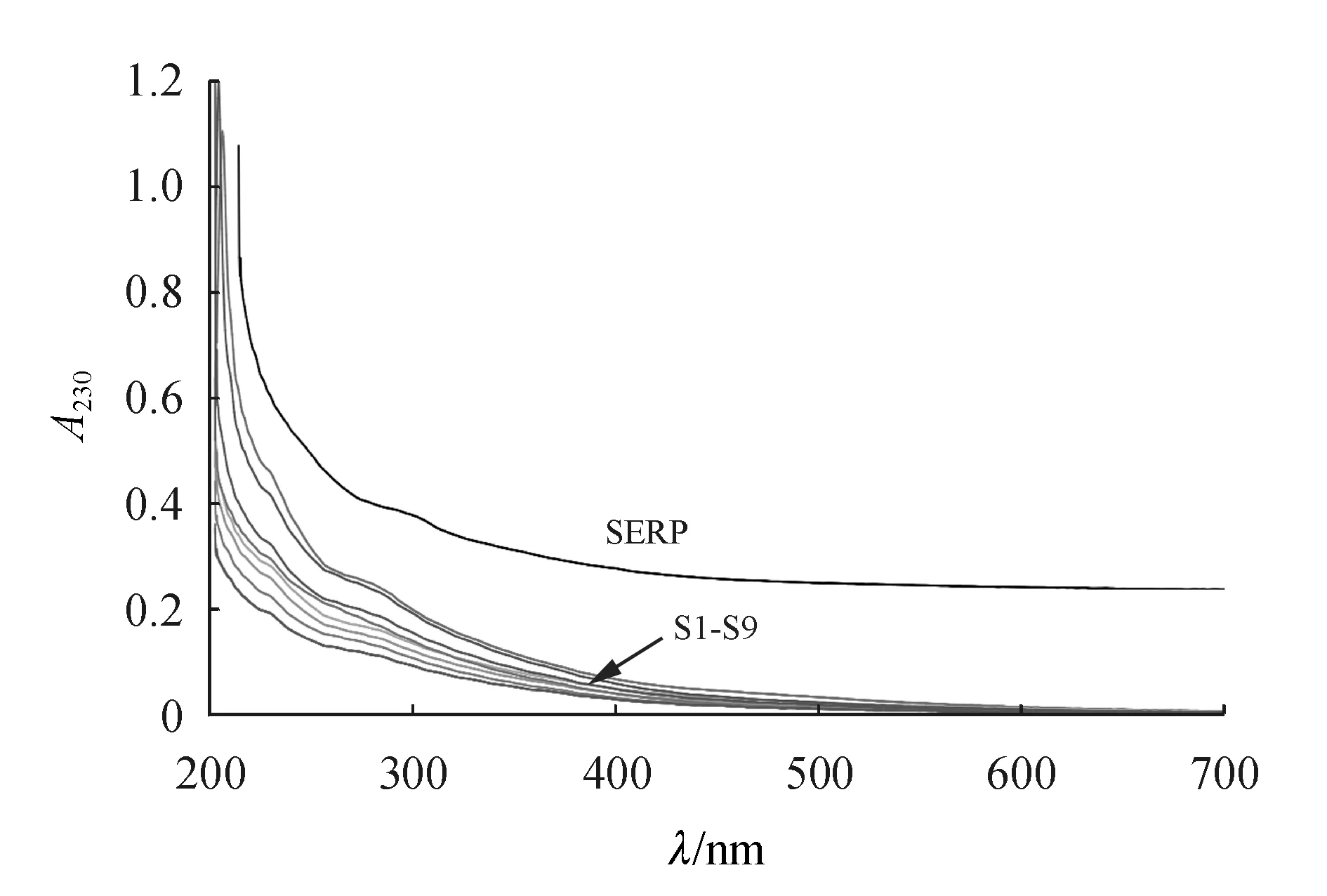
Figure3 UV spectra of SERP and S1-S9
3.6 红外光谱分析
SERP及其硒化产物的红外光谱如图4所示。在3 200~3 600 cm-1出现的宽而强的吸收峰为-OH的伸缩振动峰。2 935.2 cm-1弱的吸收带归属于C-H伸缩振动峰。1 618.5 cm-1的吸收峰为C=O伸缩振动,出现在1 280.6,1 334.5,1 422.3 cm-1的宽而强的吸收带为C-H的弯曲振动。950和1 200 cm-1之间的吸收区为碳水化合物指纹区[13]。与原糖的红外光谱相比较,9种硒化产物的红外光谱在833.6 cm-1呈现硒酸酯的Se=O伸缩振动特征吸收。同时,575.9 cm-1的特征吸收峰归属于Se-O-C的伸缩振动,这些特征峰均说明粗品多糖发生了硒化反应[14]。
3.7 热重分析
SERP及其9种硒化产物的热重分析如图5所示,在加热过程中,50~150 ℃发生的失重为多糖中结合水的部分,150 ℃之后,样品发生了分解[15]。如图所示,与SERP相比,硒化产物更容易分解,该结果表明硒的引入降低了SERP的热稳定性。

Figure4 Infrared spectra of SERP (A) and S1-S9 (B)

Figure5 TGA curves of SERP and S1-S9
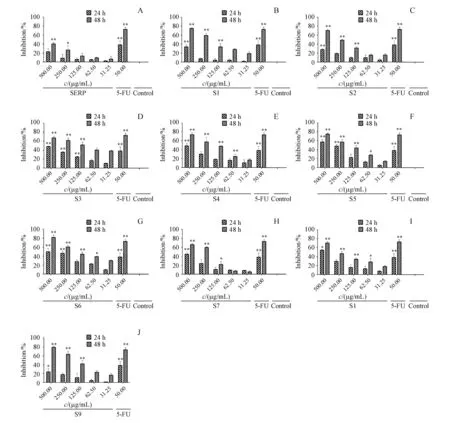
3.8 抗肿瘤活性分析
SERP及其9种硒化产物对于HepG2作用24和48 h时的抑制效果如图6所示。作用48 h时,氟尿嘧啶(50 μg/mL)对HepG2的抑制率为(71.02±5.86)%,在500 μg/mL的质量浓度下,SERP对该细胞的抑制率为(40.28±3.36)%,而9种硒化产物中最高抑制率可达到(81.99±5.29)%。与SERP相比,9种硒化多糖对于HepG2肿瘤细胞的抑制效果更为显著,其中S3的半数抑制率甚至达到(113.03±43.01) μg/mL,因此也对该产物作用于HT-29人结肠癌细胞、HeLa人宫颈癌细胞和L02人正常肝细胞的抑制效果进行了考察。结果(图7)表明,硒化肿节风浸膏残渣多糖不仅对于HepG2细胞具有抑制作用,同时对HT-29细胞、HeLa细胞同样具有抑制作用,SERP经过硒化修饰后,抗肿瘤活性得到明显提高,而对人正常肝细胞没有影响。相比于氟尿嘧啶等化学药物,该硒化多糖的毒性更低,后续可通过进一步的体内实验研究后,作为潜在的抗肿瘤药物进一步开发。
4 讨 论
本研究使用单因素与正交实验对SERP的硒化条件进行优化,最佳制备工艺为:反应温度70 ℃、反应时间7 h、HNO3质量分数1.0%、BaCl2用量1.5 g。在此条件下,硒化肿节风浸膏残渣多糖的总糖含量达到(92.30±3.22)%,硒含量为(929.77±12.51) μg/g,产率为(22.15±0.66)%。
目前对于硒化多糖的结构表征多采用紫外光谱分析,红外光谱分析和热重分析[4]。本研究对制备所得的硒化肿节风浸膏残渣多糖使用紫外光谱、红外光谱和热重分析,表明硒元素以亚硒酸酯的结构与多糖相结合。
抗肿瘤活性作为硒化多糖的常见活性,本研究对最佳条件下制备所得的硒化肿节风浸膏残渣多糖进行了抗肿瘤活性研究,在最佳条件下所制备的硒化肿节风浸膏残渣多糖对于HepG2人肝癌细胞的半数抑制率达到(123.33±23.02) μg/mL,对于HT-29人结肠癌细胞、HeLa人宫颈癌细胞也表现一定的抑制作用,而对于正常人肝细胞L02的毒性较小。与SERP相比,硒化肿节风浸膏残渣多糖具有更高的抗肿瘤活性,但其体内抗肿瘤活性还有待进一步研究,但硒化肿节风浸膏残渣多糖仍可作为抗肿瘤药物继续进行开发,为肿节风浸膏残渣的生物资源再利用提供了新思路。
*P<0.05;**P<0.01vscontrol group

*P<0.05;**P<0.01vscontrol group
[1] Chen W,Chen J,Wu H,etal.Optimization of selenylation conditions for a pectic polysaccharide and its structural characteristic[J].IntJBiolMacromol,2014,69:244-251.
[2] Qin T,Chen J,Wang D,etal.Selenylation modification can enhance immune-enhancing activity of Chinese angelica polysaccharide[J].CarbohydrPolym,2013,95(1):183-187.
[3] Sun SF,Pan LL,Wang CM,etal.Interaction of an organic selenium compound with human serum albumin[J].BiolTraceElemRes,2006,114(1):301-311.
[4] Wang J,Zhao B,Wang X,etal.Synthesis of selenium-containing polysaccharides and evaluation of antioxidant activityinvitro[J].IntJBiolMacromol,2012,51(5):987-991.
[5] Braga HC,Wouters AD,Zerillo FB,etal.Synthesis of seleno-carbohydrates derived from D-galactose[J].CarbohydrPolym,2010,345(16):2328-2333.
[6] Zhu YS,Xu W,Shao R,etal.Optimization of microwave extraction condition ofGynuradivaricatapolysaccharide by response surface analysis[J].JChinaPharmUniv(中国药科大学学报),2016,47(3):359-362.
[7] Payne RL,Southern LL.Comparison of inorganic and organic selenium sources for broilers[J].PoultSci,2005,84(84):898-902.
[8] Wang Y,Chen J,Zhang D,etal.Tumoricidal effects of a selenium (Se)-polysaccharide from Ziyang green tea on human osteosarcoma U-2 OS cells[J].CarbohydrPolym,2013,98(1):1186-1190.
[9] Maseko T,Callahan DL,Dunshea FR,etal.Chemical characterisation and speciation of organic selenium in cultivated selenium-enrichedAgaricusbisporus[J].FoodChem,2013,141(4):3681-3687.
[10] Ahn SJ,Koketsu M,Ishihara H,etal.Regulation of melanin synthesis by selenium-containing carbohydrates[J].ChemPharmBull(Tokyo),2006,54(3):281-286.
[11] Shang LC,Wu SW,Zhang C,etal.Preparation,characterization and activity analysis of selenium-containing pumpkin polysaccharide[J].FoodSci(食品科学),2016,37(19):48-53.
[12] Dubois M,Gilles K,Hamilton JK,etal.A colorimetric method for the determination of sugars[J].Nature,1951,168(4265):167.
[13] Blumenkrantz N,Asboe-Hansen G.A quick and specific assay for hydroxyproline[J].AnalBiochem,1973,55(1):288-291.
[14] Liu Z,Sun H,Shen S,etal.Simultaneous determination of total arsenic and total selenium in Chinese medicinal herbs by hydride generation atomic fluorescence spectrometry in tartaric acid medium[J].AnalChimActa,2005,550(1/2):151-155.
[15] Cui SW,Phillips GO,Blackwell B,etal.Characterisation and properties ofAcaciasenegal,(L.) Willd.var.senegal,with enhanced properties (Acacia ( sen ) SUPERGUMTM):Part 4.Spectroscopic characterisation ofAcaciasenegal,var.senegal,andAcacia(sen) SUPERGUMTMarabic[J].FoodHydrocoll,2007,21(3):347-352.
[16] Zhao B,Zhang J,Yao J,etal.Selenylation modification can enhance antioxidant activity ofPotentillaanserinaL.polysaccharide[J].IntJBiolMacromol,2013,58(2):320-328.
