基于RNA-seq数据的小麦条锈菌SSR标记开发
2018-03-14刘秀峰许静杨袁文娅时晓伟
刘秀峰,许静杨,袁文娅,梁 丹,时晓伟
(1.天津市农作物研究所,天津300381;2.天津市植物保护研究所,天津300381)
由条形柄锈菌小麦专化型(Puccinia striiformis f.sp.tritici,Pst)引起的小麦条锈病暴发性强、发生范围广,在我国曾发生多次大流行,危害严重[1]。利用分子手段研究小麦条锈菌的群体遗传结构、远距离传播等有助于了解其流行动态。简单重复序列(Simple sequence repeat,SSR)通常是由1~6 个核苷酸组成的串联重复单元,广泛分布在真核和原核生物基因组中,具有多态性高、重复性高、覆盖面广等优点。在小麦条锈菌研究中,SSR标记已应用于群体遗传多样性分析和分子标记开发等方面[2-5]。目前小麦条锈菌研究所用的SSR标记数量有限,主要来自于cDNA文库和表达序列标签(Expressed sequence tag,EST)文库[6-8]。最近报道了由小麦条锈菌基因组开发的SSR引物,即根据北美的PST130基因组序列开发的 25个 SSR引物[9]和中国的CY32基因组开发的20个多态性SSR引物[10]。由于小麦条锈菌表现出较低的遗传差异,应用SSR引物分析小麦条锈菌群体遗传结构时,往往获得的遗传差异信息过少[11]。可见,现有的小麦条锈菌SSR标记数量不能满足其分子生物学研究的需求。为了提高SSR分辨小麦条锈菌遗传差异的能力,有必要开发更多的SSR标记。RNA-seq是近年建立的基于深度测序的转录组分析新技术。本研究在通过RNA-seq技术对小麦条锈菌夏孢子形成时转录组测定基础上,分析其转录组序列,发掘SSR位点,旨在为小麦条锈菌群体遗传结构分析等开发新的SSR引物。
1 材料和方法
1.1 材料
小麦条锈菌为CY31、CY32、CY33小种和致病型V26单孢子堆分离物,用于繁殖的小麦品种为铭贤169。
1.2 方法
1.2.1 小麦条锈菌采集、RNA提取及测序 将小麦条锈菌分离物分别制成孢子悬浮液,用微型喷雾器分别喷雾接种于铭贤169第1片完全展开叶上。10℃下黑暗保湿 24 h后,放置于人工气候室内[17℃/14℃(日/夜),每日光照16 h]。待叶片出现花斑后,剪去多余心叶,加上隔离罩。接种后16 d开始产生夏孢子时,立即分别收集夏孢子保存于液氮中。分别称取20 mg小麦条锈菌夏孢子,采用Trizol法(Invitrogen,USA)提取小麦条锈菌总RNA,形成该时刻转录组的RNA池。提取过程中所用器具和耗材均经过DEPC处理。采用琼脂糖凝胶电泳检测RNA完整性,并用微量紫外分光光度计检测其纯度和浓度,然后利用Illumina HiSeqTM2000进行转录组测序,由诺禾致源公司执行。
1.2.2 小麦条锈菌转录组SSR分析 对转录组测序得到的原始reads进行质量控制,去除带接头的reads,去除片段内未知碱基比例大于10%的reads,去除质量值小于20的碱基数占整个reads 50%以上的低质量 reads。然后,用 TopHat2[12]将过滤后的reads比对到小麦条锈菌基因组上,其错配碱基数不高于2 bp。使用Trinity软件[13]将未比对到小麦条锈菌基因组的reads进行组装,参数为默认参数,最短contig长度设置为200 bp。比对和组装以后得到小麦条锈菌转录组的转录本,选取每个基因最长的转录本作为unigenes。将4个小麦条锈菌分离物转录组的unigenes合并,去除重复的unigenes,得到小麦条锈菌转录组高质量的unigenes。
利用 MISA 软件参照 http://pgrc.ipkgatersleben.de/misa/misa.html中步骤对小麦条锈菌unigenes进行SSR位点搜索,搜索的SSR基序重复单元长度为1~6个核苷酸,其限制条件为单核苷酸重复不低于10次,二核苷酸重复不低于6次,三核苷酸、四核苷酸、五核苷酸和六核苷酸重复不低于5次,2个SSR位点间的距离不大于100 bp则视为复合 SSR。用 Primer 3(http://primer3.sourcegorge.net)软件对搜索到的SSR位点设计引物。设计原则为引物序列中不含SSR、获得的引物序列可以比对到unigenes序列、去除可比对到不同unigenes序列的引物、引物的5'端允许有3个碱基的错配、退火温度在55~64℃、引物长度在18~27 bp、G+C含量在40%~60%。同时注意尽量避免出现发卡结构等二级结构。
随机合成100对符合筛选条件的引物,每对引物中一条引物的5'端连接FAM荧光素,内标为GS500LIZ。以小麦条锈菌 CY31、CY32、CY33基因组DNA为模板进行扩增,PCR产物经ABI 3730XL分析后进行引物筛选。
2 结果与分析
2.1 小麦条锈菌RNA-seq数据质量评估
小麦条锈菌CY31、CY32、CY33小种和致病型V26的 RNA 质量浓度范围在 165~206 ng/μL,18S、28S条带清晰(图1)。4个样品共产生396 711 824个原始reads,原始数据每种碱基含量约为25%,说明原始数据质量较高。经过质量控制后得到382 125 418个reads。将过滤后reads与小麦条锈菌基因组(PST-78,GenBank:AJIL00000000.1)比对,结果显示,4个样品数据的 71.15%~74.21%比对到了参考基因组上,其中单个定位的测序序列(Uniquely mapped reads)占总体(Clean reads)的百分比在70.14%~73.13%,多个定位的测序序列(Multiple mapped reads)占总体的百分比在 0.99%~1.16%。这些结果说明本研究所使用的测序数据真实可靠,能够保证分析结果的正确性。

图1 小麦条锈菌RNA
2.2 小麦条锈菌夏孢子形成时转录组的SSR分布
4个小麦条锈菌分离物转录组57.32 Gb的clean reads组装unigenes后,合并、去除重复的unigenes得到20 802条高质量unigenes,总碱基数为27 893 297 bp,平均每条unigene长约1 340 bp。根据1.2.2中所述条件进行SSR筛选,在2 941条unigenes中发现6 083个SSR位点。SSR发生频率(含有SSR的unigenes数目占总unigenes数目的比例)为14.13%,平均每9.84 kb出现1个SSR 位点,其中包含单个SSR位点的unigenes有1 615条,占含有SSR位点 unigenes总条数的 54.9%。1 326条unigenes含有1个以上SSR位点,其中绝大多数含有2~4个 SSR位点,6条 unigenes含有12个 SSR位点,含有14、15、20个SSR位点的unigenes各1条。
小麦条锈菌夏孢子形成时转录组SSR重复类型丰富,从单核苷酸重复到六核苷酸重复均有出现。6种重复核苷酸类型组成的SSR数量存在较大差异,以单核苷酸重复(2 575个)和三核苷酸重复(2 427个)所占比例较高,分别占SSR总数的42.33%和39.90%,分布频率(SSR数量/总unigenes数量)分别为12.38%和11.67%。总体上,二、四、五、六核苷酸重复类型的SSR数量依次递减,但六核苷酸重复类型的SSR数量大于五核苷酸重复类型的SSR数量。11个五核苷酸重复类型的SSR数量均为1个(表1)。
小麦条锈菌转录组中含有丰富的重复基序,不同重复类型的小麦条锈菌转录组SSR中有多种基序。单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸重复SSR出现的基序种类分别为4、10、56、44、11、19 种,共有 144 种。在本研究筛选的SSR中,单核苷酸重复的优势基序为T,占单核苷酸重复的60.00%,G重复类型所占比例最少。在三核苷酸重复基序中,优势基序有AAC、ATC、CAT,分别为301、140、138个,AAC重复类型的SSR所占比例最高,而TTG类型的SSR数量最少,仅有1个。五核苷酸和六核苷酸重复的SSR数量较少,二者加起来仅占 SSR总数的 0.53%,分布频率分别为0.05%和 0.10%(表 1)。
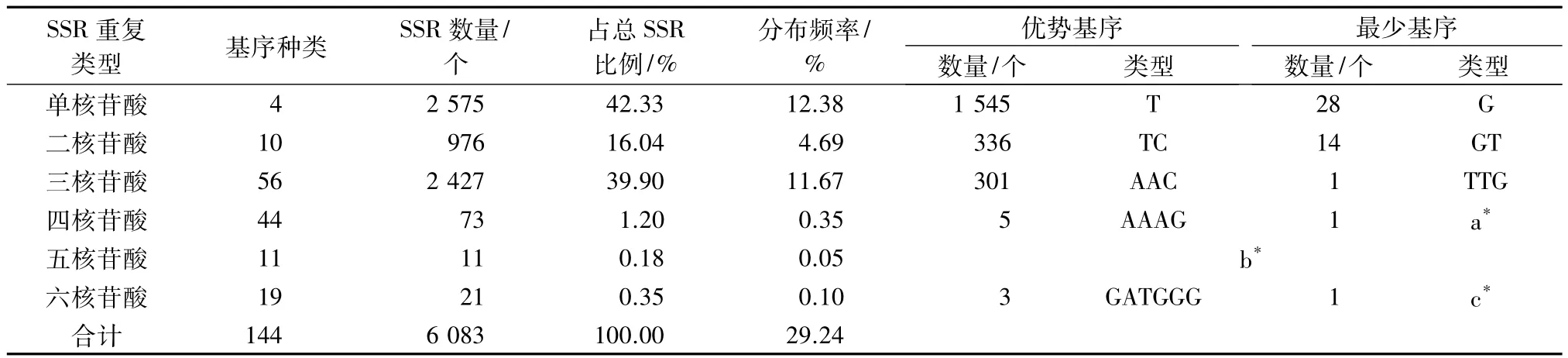
表1 小麦条锈菌夏孢子形成时转录组SSR重复类型分布特征
2.3 小麦条锈菌夏孢子形成时转录组SSR重复次数和基序长度
从重复次数上看,小麦条锈菌转录组SSR各重复类型的重复次数存在明显差异。除单核苷酸外,5、6次重复的SSR最多,分别为1 488个和1 016个,两者之和占总SSR个数的41.16%,7次重复的SSR有452个,占 7.43%,≥12次重复的 SSR个数为63个,所占比例仅为1.04%。表2中数据表明,随着重复次数的增加各重复类型SSR数量呈现出递减的趋势。二核苷酸重复的主要重复数为6~8次,三核苷酸重复的主要重复数为5~8次,四核苷酸重复的主要重复数为5~6次,五、六核苷酸重复的主要重复数为5次,说明随着核苷酸重复数的增加,同一重复类型的SSR总数也呈现变小的趋势。可见,基序重复次数的变异引起位点长度的变异是产生SSR多态性的主要原因。另外,分析数据表明,基序连续重复数最多的为三核苷酸 TAC,达 32次(unigene为PSTG-07600),T基序连续重复可达31次(unigene为PSTG-16488)。六核苷酸基序的连续重复次数整体上大于五核苷酸,其中AAGGAG基序连续重复次数为15次(unigene为 PSTG-04191),AAAACA(unigene为 PSTG-16928)和 CTCAAT(unigene为PSTG-14376)基序连续重复次数均为11次,而五核苷酸基序的连续重复次数最多为6次。

表2 小麦条锈菌夏孢子形成时转录组SSR各重复基序长度的重复次数分布特征 个
统计SSR长度分布发现,小麦条锈菌转录组SSR基序长度分布在10~435 bp,其中复合SSR的长度在22~435 bp,主要集中在 80~199 bp,共有934个,占总SSR个数的15.35%。除去复合 SSR,总体上小麦条锈菌转录组SSR基序长度分布在10~90 bp,12~20 bp基序长度的SSR数量最多(3 238个),占总SSR个数的53.22%,大部分基序长度集中在 15 bp,10~11 bp的 SSR 有 1 390个,占 22.85%。大于20 bp的SSR数量较少,有521个,占8.6%(图2)。

图2 小麦条锈菌夏孢子形成时转录组SSR基序长度分布
2.4 小麦条锈菌夏孢子形成时转录组SSR引物筛选
选取 100对荧光引物对小麦条锈菌 CY31、CY32、CY33基因组 DNA进行 PCR扩增,ABI 3730XL检测结果显示,共有87对引物产生清晰、稳定的峰,即均有扩增产物。其中,75对引物扩增产物显示1个峰,12对引物显示2~4个峰,即2~4个等位基因。
3 结论与讨论
SSR标记传统开发方法是构建cDNA文库筛选,高通量测序技术为大规模筛选SSR标记提供了新的工具,例如从小麦条锈菌PST130基因组中发现1 889个SSR位点[9]。转录组是在特定的发育阶段和不同的生理条件下所有转录的RNA集合。利用转录组数据获得含有微卫星的序列,并用其进行遗传多样性研究在一些物种上已有报道。本研究利用小麦条锈菌夏孢子形成时的转录组数据大规模开发SSR标记位点,获得的SSR位点中除单核苷酸外,以三核苷酸重复基序为主要类型,与由小麦条锈菌基因组发掘的SSR位点中的主导类型为二、三核苷酸重复基序的结果一致[10]。而且,目前报道的小麦条锈菌SSR中的一部分与本研究发掘的SSR位点信息完全相同或相近。例如,Zhan等[10]报道的WSR7、WSR23、WSR90与本研究中相应SSR的重复基序、重复次数以及设计的引物对完全一致,WSR79、WSR95、WSR62、WSR85 等与本研究中相应SSR重复基序一致,与设计引物对的其中1条一致,说明利用小麦条锈菌转录组开发SSR引物是可行的。随机合成的100对引物中,有87对可以扩增出产物,说明设计的引物有效,在后续工作中需要加强开发在小麦条锈菌群体中具有多态性的引物。本研究设计的引物合成时进行了荧光修饰,该方法虽然费用比琼脂糖凝胶电泳检测略高,但筛选效率高,工作量相对较小,适合大规模筛选SSR引物。利用转录组开发的SSR位点可能与功能基因相关,由小麦条锈菌转录组开发的SSR中的一部分与由基因组发掘的SSR具有一致性,可能与该位点基因在小麦条锈菌中具有保守性有关。最近报道了小麦条锈菌中一个高度保守的效应子Puccinia NPR1 interactor(PNPi),推测PNPi的高度保守性与小麦条锈菌在进化竞争中取得优势有关,改变PNPi蛋白结构可能影响小麦条锈菌的寄生适合度[14]。
通过分析小麦条锈菌夏孢子形成时转录组数据,配合荧光标记,可以批量筛选SSR引物,有望发现小麦条锈菌产孢过程中表达基因的等位位点变异,设计出更多的具多态性的SSR引物,还可能发现与小麦条锈菌产孢量密切相关的基因。目前,笔者正在利用多个小麦条锈菌分离物筛选本研究设计的SSR引物,筛选出的多态性引物加上已经报道的SSR引物可以应用在大范围的小麦条锈菌基因型和群体动态研究中。
[1] 李振岐,曾士迈.中国小麦锈病[M].北京:中国农业出版社,2002.
[2] Shan W X,Chen S Y,Kang Z S,et al.Genetic diversity in Puccinia striiformis Westend.f.sp.tritici revealed by pathogen genome-specific repetitive sequence[J].Can J Botany,1998,76(4):587-595.
[3] Enjalbert J,Duan X Y,Leconte M,et al.Genetic evidence of localadaptation ofwheatyellow rust(Puccinia striiformis f.sp.tritici)within France[J].Mol Ecol,2005,14(7):2065-2073.
[4] 陆宁海,郑文明,王建锋,等.陇南地区小麦条锈菌群体遗传多样性 SSR分析[J].中国农业科学,2009,42(8):2763-2770.
[5] 代君丽,刘珂,于思勤,等.河南省小麦条锈菌特异性SSR 标记的筛选[J].麦类作物学报,2011,31(1):166-169.
[6] Chen C Q,Zheng W M,Buchenauer H,et al.Isolation of microsatellite loci from expressed sequence tag library of Puccinia striiformis f.sp.tritici[J].Mol Ecol Resour,2009,9(1):236-238.
[7] Enjalbert J,Duan X Y,Vautrin D,et al.Isolation of twelve microsatellite loci,using an enrichment protocol,in the phytopathogenic fungus Puccinia striiformis f.sp.tritici[J].Mol Ecol Notes,2002,2(4):563-565.
[8] Bahri B,Leconte M,De Vallavieille-Pope C,et al.Isolation of ten microsatellite loci in an EST library of the phytopathogenic fungus Puccinia striiformis f.sp.tritici[J].Conserv Genet,2009,10(5):1425-1428.
[9] Bailey J,Karaoglu H,Wellings C R,et al.Isolation and characterization of 25 genome-derived simple sequence repeat markers for Puccinia striiformis f.sp.tritici[J].Mol Ecol Resour,2013,13(4):760-762.
[10] Zhan G M,Wang F P,Luo H Y,et al.Screening for simple sequence repeat markers in Puccinia striiformis tritici based on genomic sequence[J].J Zhejiang Univ Sci B,2015,16(8):727-732.
[11] Loladze A.Pathogen and host variability studies in wheat stripe rust in Australia[D].Sydney:University of Sydney,2010.
[12] Kim D,Pertea G,Trapnell C,et al.TopHat2:Accurate alignment of transcriptomes in the presence of insertions,deletions and gene fusions[J].Genome Biol,2013,14(4):R36.
[13] Grabherr M G,Haas B J,Yassour M,et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J].Nat Biotechnol,2011,29(7):644-652.
[14] Wang X D,Yang B J,Li K,et al.A conserved Puccinia striiformis protein interacts with wheat NPR1 and reduces induction of pathogenesis-related genes in response to pathogens[J].Mol Plant Microbe Interact,2016,29(12):977-989.
