HPLC-MS/MS检测血浆百草枯浓度方法的建立
2018-03-07岳晓静李平法
岳晓静, 李 卫, 李平法
(1.焦作市妇幼保健院检验科,河南 焦作 454000;2.焦作市第一人民医院检验科,河南 焦作 454000;3.新乡医学院医学检验学院,河南 新乡 453004)
百草枯是一种有效成分为l,1-二甲基-4,4-联吡啶阳离子盐的除草剂,被广泛用于农业生产中[1]。由于百草枯对环境、水的污染作用,且对人体的毒性作用很强,因此在20世纪80年代后期,许多国家已经开始禁用。然而,我国和东南亚地区目前仍在使用百草枯,且中毒现象仍很常见,每年都有很多百草枯中毒死亡案例[2-4]。百草枯中毒死亡率为60%~70%[5],目前尚无特效药可以解毒。有研究表明,百草枯中毒的死亡率与血中百草枯浓度高度相关[6-7],因此其浓度是临床诊断百草枯中毒最重要的指标[8-9]。一般来说,如果患者血中百草枯浓度在中毒后4、6、10、24 h分别高于2 000、600、300和100 ng/mL,其生存的机率将非常低[10]。因此,早期快速、准确地测定血中百草枯浓度至关重要。目前,用于百草枯检测的方法有光谱法和色谱法,但稳定性不能满足要求,且操作复杂繁琐。质谱法具有高选择性和高敏感性的优点,其在临床上的应用已受到了广泛关注。本研究拟建立高效液相色谱串联质谱(high performance liquid chromatographytandem mass spectrometry,HPLC-MS/MS)检测血中百草枯浓度的方法,为临床早期诊断百草枯中毒提供帮助。
1 材料和方法
1.1 研究对象
选取2015年7月—2016年6月在新乡医学院第一附属医院急诊科住院治疗的百草枯急性中毒患者75例,其中男37例、女38例,年龄16~62岁,均为口服20%百草枯溶液中毒患者(均以自杀为目的),于服药后2~6 h入院,入院后采用常规治疗(如洗胃、抗氧化剂治疗、激素治疗、血液灌流等)。排除既往有严重肺部疾病或心、肝、肾功能不全者,伴有其他药物中毒者,入院放弃治疗或30 d内失访者以及资料信息不全者。采集所有患者入院后30 min内的血液样本,以乙二胺四乙酸二钾(ethylenediaminetetraacetic acid-K2,EDTAK2)抗凝, 2 200×g离心5 min,分离血浆,保存于-80 ℃。根据入院30 d后是否死亡分为存活组(22例)和死亡组(53例)。2个组之间性别、年龄、中毒至就诊的时间差异均无统计学意义(P>0.05)。
1.2 仪器和材料
LC-20A高效液相色谱仪(日本岛津公司),API 4000三重四级杆质谱仪(美国爱博才思公司),PAL-CAXXBIOM2型超纯水仪(美国颇尔公司),AUW2201型分析天平(日本岛津公司),UGC-24CE型氮吹仪(北京优晟联合科技有限公司),TGL-16M型高速离心机(湖南赛特湘仪离心机仪器有限公司),OF1型旋涡混合器(上海琪特分析仪器有限公司)。百草枯标准品(批号:MM40TC28)、内标乙基百草枯标准品(批号:DD40TC99)购自上海子起生物科技有限公司,甲醇(色谱级,美国飞世尔公司)、甲酸铵(色谱级)购自天津市科密欧化学试剂有限公司;超纯水为自制;空白血液(健康献血者血液)购自新乡市中心血站。
1.3 方法
1.3.1 仪器设置 百草枯及其内标的质谱条件见表1。选择电喷雾电离(electrospray ionization,ESI)源及正离子条件下多离子反应监测(multiple reaction monitoring,MRM)扫描模式。色谱条件:Phenomenex Kinetex 2.6 μm HILIC色谱柱(100.0 mm×2.1 mm),以1%甲酸水溶液(含250 mmol/L甲酸铵)为流动相A、乙腈为流动相B,梯度洗脱(0.01 min→0.30 min,45% B→35%B;0.30 min→4.00 min,35% B;4.00 min→5.00 min,35% B→45% B;5.00 min→6.50 min,45% B),流速为0.35 mL/min,进样量为10 μL,柱温为40 ℃。
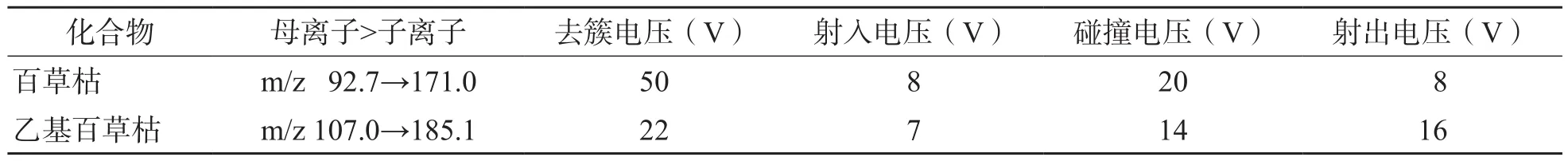
表1 百草枯及其内标乙基百草枯的质谱条件
1.3.2 百草枯及其内标乙基百草枯工作液的制备方法 称取26.38 mg百草枯标准品,用甲醇于100 mL容量瓶中定容,从中吸取5 mL于100 mL容量瓶中,用甲醇定容,得浓度为13 190 ng/mL的百草枯工作液。称取33.66 mg乙基百草枯标准品,用甲醇于100 mL的容量瓶中定容,从中吸取1 mL于100 mL容量瓶中,用甲醇定容,得浓度为3 366 ng/mL的乙基百草枯工作液。
1.3.3 百草枯标准液分析制备方法 取1.5 mL Eppendorf管,吸取对应编号的百草枯标准液各100 μL,加入Eppendorf管底部,再加入100 μL空白血浆,然后加入50 μL内标(3 366 ng/mL乙基百草枯)和350 μL甲醇,密封,漩涡30 s,室温(25 ℃)下22 560×g离心10 min。分2次,每次吸取200 μL上清液于1.5 mL进样小瓶中,再加入200 μL超纯水,混匀后待测。
1.3.4 样本处理 取100 μL血浆置于1.5 mL Eppendorf管中,吸入50 μL内标(3 366 ng/mL乙基百草枯)和450 μL甲醇,22 560×g高速离心10 min,沉淀蛋白。而后分2次,每次吸取200 μL于1.5 mL进样小瓶中,加入200 μL超纯水,混匀后采用HPLC-MS/MS检测。根据百草枯峰面积与内标乙基百草枯峰面积的相对值计算血浆百草枯浓度。
1.4 方法学评价
1.4.1 百草枯标准曲线绘制及线性评价 取百草枯工作液适量,逐级稀释配成6种浓度:13 190.00、8 793.00、4 397.00、1 467.00、489.00、54.28 ng/mL。采用HPLC-MS/MS重复检测3次。以百草枯浓度为X轴,百草枯峰面积与内标峰面积的比值为Y轴绘图,观察其线性情况。
1.4.2 准确度和精密度评价 将高、中、低及定量下限4种浓度标准液按标准液分析制备方法处理。高浓度(9 893 ng/mL)的配制方法:取1 500μL百草枯工作液,加500 μL甲醇,混匀;中浓度(6 595 ng/mL)的配制方法:取500 μL百草枯工作液,加500 μL甲醇,混匀。低浓度(163 ng/mL)的配制方法:取489 ng/mL的百草枯溶液1 mL,加2 mL甲醇,混匀。定量下限浓度为54.28 ng/mL。各浓度标准液平行制备5份,进样分析来考察批内准确度和精密度,每个浓度每天检测1次,共检测3 d,以此观察批间准确度和精密度。
1.4.3 选择性和基质效应评价 通过计算6份空白血浆响应值与定量下限响应值均值的比值以及6份空白血浆响应值与内标响应值均值的比值来观察方法的选择性。通过计算6批空白基质的经内标归一化的基质因子的变异系数来考察基质效应(在高浓度和低浓度下进行)。
1.4.4 稳定性评价 分别观察高、低浓度样本处理后的室温放置稳定性(室温放置2 h、8 h及1 d后检测)、冻融稳定性(样本置于-80 ℃冰箱中冻存,然后室温融化,重复冻融1、2及3次)以及长期存储稳定性(分别置于-80 ℃冰箱中冻存1个月和放置2~8 ℃保存1周,然后室温融化或放置到室温)。
1.5 统计学方法
采用SPSS 17.0软件进行统计分析。计量资料呈非正态分布,采用中位数(范围)表示,组间比较采用Mann-Whitney U检验。采用受试者工作特征(receiver operating characteristic,ROC)曲线评估血浆百草枯浓度对临床结局的判断价值。以P<0.05为差异有统计学意义。
2 结果
2.1 线性评价
百草枯浓度在54.28~13 190.00 ng/mL范围内线性关系良好,回归方程为Y=0.000 1X+0.011 6,r2=0.998 3。HPLC-MS/MS检测百草枯的标准曲线见图1。
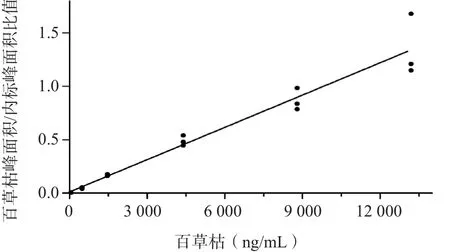
图1 HPLC-MS/MS检测百草枯的标准曲线
2.2 准确度和精密度评价
当百草枯浓度处于定量下限时,批内准确度为96.31%~120.04%,批间准确度为104.43%;高、中、低浓度批内和批间检测值与理论值的偏差均在±15%范围内,符合要求。高、中、低浓度及定量下限的批内和批间相对标准偏差(relative standard deviation,RSD)均 <15%,符合要求(定量下限处的RSD<20%)。见表2。
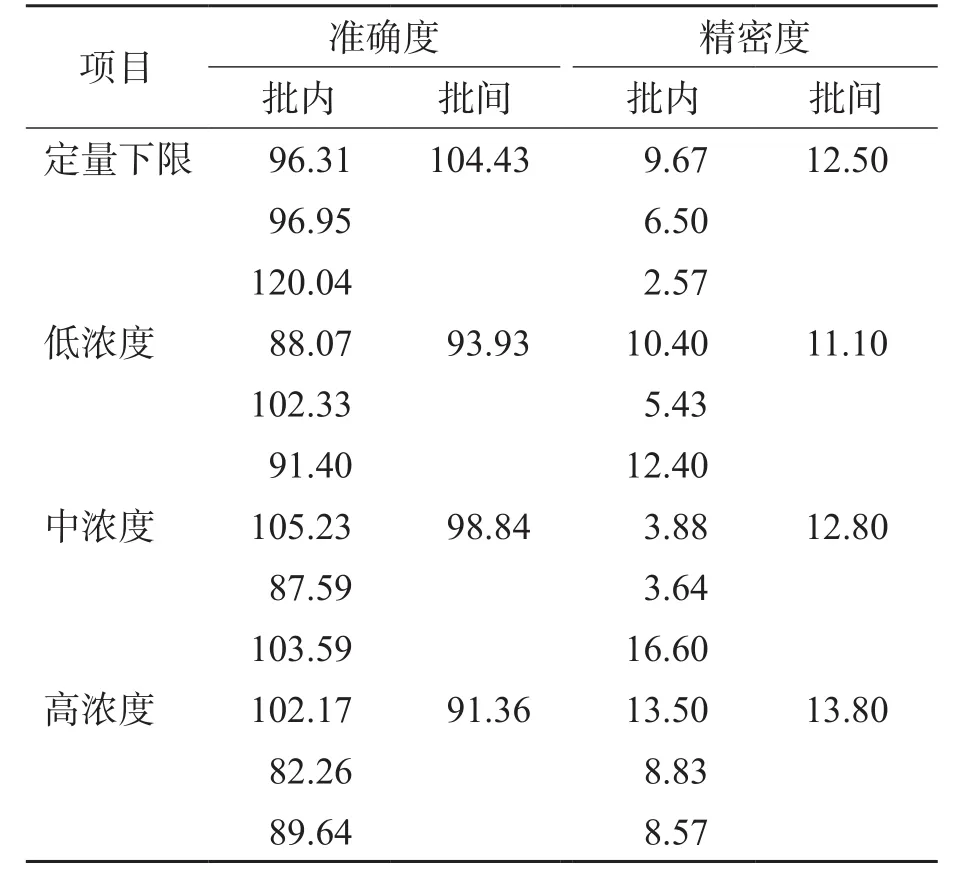
表2 HPLC-MS/MS测定百草枯的准确度和精密度(%)
2.3 选择性和基质效应评价
采用HPLC-MS/MS测定6份空白血浆,结果显示百草枯对应保留时间均无干扰峰,响应值为零,选择性符合要求。百草枯标准品、空白样本内标、空白样本百草枯、内标标准品的选择离子流图见图2~图5。高、低浓度计算得到的内标归一化的基质因子变异系数分别为7.53%、13.3%,符合要求,见表3。
图2 百草枯标准品选择离子流图
图3 空白样本内标选择离子流图
图4 空白样本百草枯选择离子流图
图5 内标选择离子流图
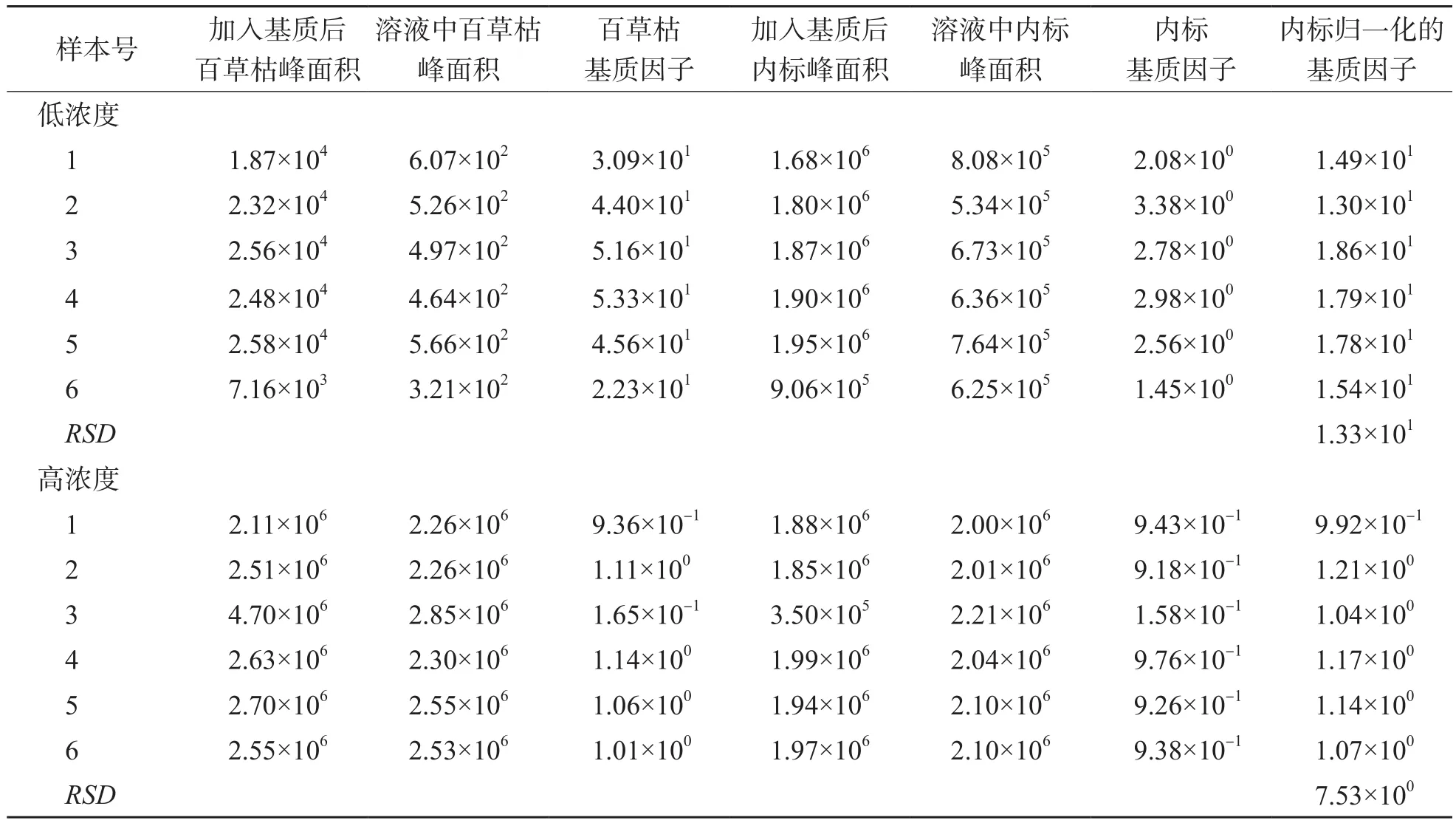
表3 HPLC-MS/MS测定百草枯的基质效应分析
2.4 稳定性评价
除高浓度样本处理后放置8 h的准确度(118.66%)略高于上限要求(115.00%)外,其他样本均符合要求。见表4。
2.5 临床验证
2.5.1 死亡组和存活组血浆百草枯浓度比较 75例百草枯中毒患者均为口服20%百草枯溶液后2~6 h采血,血浆百草枯浓度为2 820(0~22 200)ng/mL。死亡组血浆百草枯浓度为6 650(1 950~22 200)ng/mL,明显高于存活组[680(0~2 990)ng/mL](P<0.05)。
2.5.2 血浆百草枯浓度判断临床结局的ROC曲线分析 ROC曲线分析显示,血浆百草枯浓度判断百草枯中毒患者临床结局的曲线下面积为0.855,95%可信区间为0.772~0.937,P=0.000。血浆百草枯浓度越高,患者死亡的可能性越大。Youden指数最大为0.589,其对应的百草枯浓度(最佳临界值)为2 431 ng/mL,敏感性为78.9%,特异性为80.0%。见图6。
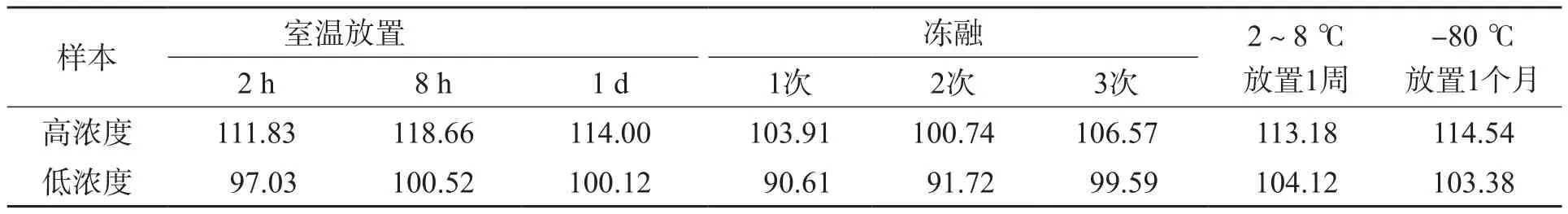
表4 HPLC-MS/MS测定百草枯浓度的稳定性评价 (%)
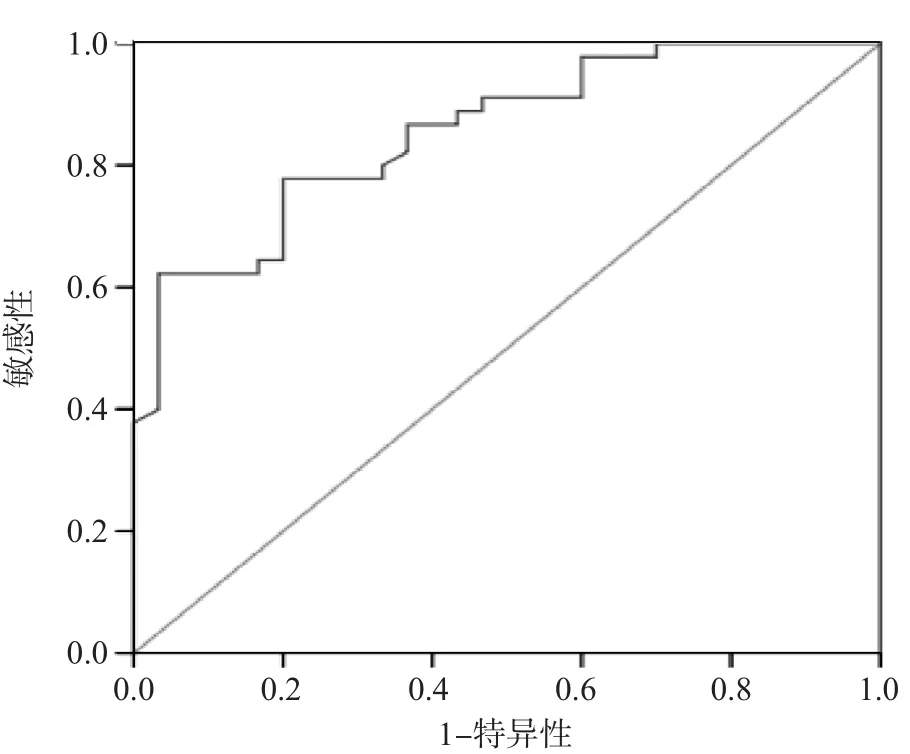
图6 血浆百草枯浓度判断临床结局的ROC曲线
3 讨论
由于百草枯极性较强,在溶液中带双重阳离子的化学特性,因此适合用HPLC-MS/MS检测。在百草枯生物样本检测中,很多色谱分析法常有繁琐的样本前处理过程,包括液-液萃取、固相萃取、微波辅助的溶剂萃取以及磁性碳纳米管萃取等[11-14],导致操作繁琐、耗时。本研究中的样本处理只需用甲醇沉淀蛋白1个步骤,简便、省时。
本研究所选用的HILIC色谱柱较普通的C18色谱柱更适合分离强极性、高水溶性的有机化合物。HILIC色谱法是串联质谱中极性化合物分析的一种替代方法[15],适合于质谱分析中反相色谱柱不保留的极性物质的分析。本研究曾选择了不同的色谱柱和流动相进行预试验:(1)选择C18色谱柱,以含250 mmol/L乙酸铵的超纯水溶液为流动相A、乙腈为流动相B,结果显示百草枯及其内标不出峰;(2)选择C18色谱柱,以含250 mmol/L乙酸铵和1.0%乙酸的超纯水溶液为流动相A、乙腈为流动相B,结果显示百草枯及其内标在色谱柱中不保留;(3)选择HILIC 100A LC色谱柱(100.0 mm×2.1 mm),以含250 mmol/L乙酸铵和1.0%乙酸的超纯水溶液为流动相A、乙腈为流动相B,结果显示百草枯及其内标在色谱柱中吸附较强,形成蘑菇峰;(4)选择HILIC 100A LC色谱柱(100.0 mm×2.1 mm),以含250 mmol/L乙酸铵和1.0%乙酸的超纯水溶液为流动相A、乙腈为流动相B,结果显示百草枯及其内标出峰较好。另外,甲酸铵的选择也是本研究所建方法的一大特点。传统的离子对试剂,如七氟丁酸[16]和三氟乙酸[11]常被用于HPLC-MS/MS分析中。然而,由于这些离子对试剂在流动相中产生离子抑制作用,使得分析灵敏度大大下降,而且给质谱系统带来额外的污染物。而本研究选择的甲酸铵能够弥补传统离子对试剂的缺陷。
本研究方法学评价结果显示,本法的线性范围为54.28~13 190.00 ng/mL,能够涵盖临床上大部分样本的浓度范围;具有较好的准确度和精密度;除定量下限处第3批平均准确度略微高于上限要求(120%)外,其余均符合要求;方法选择性好,百草枯出峰位置无干扰,基质效应符合要求;样本在-80 ℃储存可稳定1个月,由于研究时间的关系,未能观察更长时间的储存稳定性。
采用本研究建立的HPLC-MS/MS方法测定75例百草枯中毒患者的血浆百草枯浓度,结果显示死亡组血浆百草枯浓度明显高于存活组(P<0.05)。ROC曲线分析显示血浆百草枯浓度判断百草枯中毒患者临床结局的最佳临界值为2 431 ng/mL,敏感性为78.9%,特异性为80.0%,曲线下面积为0.855。本研究结果与杜宇等[17]的研究结果基本一致。由于本研究样本例数较少,HPLC-MS/MS检测血浆百草枯浓度的临床应用价值还有待今后积累更多的病例进行评价。
综上所述,本研究建立的检测血浆百草枯浓度的HPLC-MS/MS方法具有准确、可靠、特异性好、干扰少等优点,并可即时检测、30 min内出结果,因此可作为血浆百草枯浓度检测的常规方法。
[1] PATEIRO-MOURE M,ARIAS-ESTÉVEZ M,SIMAL-GÁNDARA J. Critical review on the environmental fate of quaternary ammonium herbicides in soils devoted to vineyards[J]. Environ Sci Technol,2013,47(10):4984-4998.
[2] DINIS-OLIVEIRA R J,SARMENTO A,REIS P,et a1. Acute paraquat poisoning:report of a survival case following intake of a potential lethal dose[J].Pediatr Emerg Care,2006,22(7):537-540.
[3] RAHMAN M,LEWIS D M,ALLISON K. A case of paraquat burns following an industrial accident[J].Emerg Med J,2007,24(11):777.
[4] PODPRASART V,SATAYAVIVAD J,RIENGROJPITAK S,et a1. No direct hepatotoxic potential following a multiple-low dose paraquat exposure in rat as related to its bioaccumulation[J].Toxicol Lett,2007,170(3):193-202.
[5] TAN J T,LETCHUMAN RAMANATHAN G,CHOY M P,et a1. Paraquat poisoning:experience in hospital taiping (year 2008-october 2011)[J].Med J Malaysia,2013,68(5):384-388.
[6] SCHERRMANN J M,HOUZE P,BISMUTH C,et a1. Prognostic value of plasma and urine paraquat concentration[J]. Hum Toxicol,1987,6(1):91-93.
[7] SEOK S,KIM Y H,GIL H W,et a1. The time between paraquat ingestion and a negative dithionite urine test in an independent risk factor for death and organ failure in acute paraquat intoxication[J]. J Korean Med Sci,2012,27(9):993-998.
[8] SUN L,LI G Q,YAN P B,et a1. Prediction of outcome following paraquat poisoning by arterial lactate concentration-time data[J]. Exp Ther Med,2014,8(2):652-656.
[9] YOUNES A,OKI Y,BOCIEK R G,et a1.Mocetinostat for relapsed classical Hodgkin's lymphoma:an open-label,single-arm,phase 2 trial[J]. Lancet Oncol,2011,12(13):1222-1228.
[10] PROUDFOOT A T,STEWART A T,LEVITT T,et a1. Paraquat poisoning:significance of plasmaparaquat concentrations[J]. Lancet,1979,2(8138):330-332.
[11] RUAN X L,QIU J J,WU C,et a1. Magnetic single-walled carbon nanotubes-dispersive solidphase extraction method combined with liquid chromatography-tandem mass spectrometry for the determination of paraquat in urine[J]. J Chromatogr B Analyt Technol Biomed Life Sci,2014,965:85-90.
[12] WHITEHEAD R D JR,MONTESANO M A,JAYATILAKA N K,et a1. Method for measurement of the quaternary amine compounds paraquat and diquat in human urine using high-performance liquid chromatography-tandem mass spectrometry[J]. J Chromatogr B Analyt Technol Biomed Life Sci,2010,878(27):2548-2553.
[13] BAECK S K,SHIN Y S,CHUNG H S,et a1.Comparison study of the extraction methods of paraquat in post-mortem human blood samples[J].Arch Pharm Res,2007,30(2):235-239.
[14] WINNIK B,BARR D B,THIRUCHELVAM M,et a1. Quantification of paraquat,MPTP,and MPP+in brain tissue using microwave-assisted solvent extraction(MASE) and high-performance liquid chromatography-mass spectrometry[J]. Anal Bioanal Chem,2009,395(1):195-201.
[15] BUSZEWSKI B,NOGA S. Hydrophilic interaction liquid chromatography(HILIC)-a powerful separation technique[J]. Anal Bioanal Chem,2012,402(1):231-247.
[16] WANG K C,CHEN S M,HSU J F,et al.Simultaneous detection and quantitation of highly water-soluble herbicides in serum using ion-pair liquid chromatography-tandem mass spectrometry[J].J Chromatogr B Analyt Technol Biomed Life Sci,2008,876(2):211-218.
[17] 杜宇,牟奕. 三种方法对急性百草枯中毒严重程度和预后评估价值的比较[J]. 中南大学学报(医学版),2013,38(7):737-742.
