N催化剂中活性组分的核磁共振氢谱定量研究
2018-03-06黄文氢
殷 杰,黄文氢
(中国石油化工股份有限公司 北京化工研究院,北京 100013)
从第一代Ziegler-Natta (Z-N) 催化剂发展到目前的高效载体型催化体系,作为调节剂的给电子体是催化剂研究的核心[1]。给电子体化合物在Z-N催化剂中的作用主要有:使无规活性中心失活,使无规活性中心转化为等规活性中心,以及提高等规活性中心的链增长常数等。按照加入方式的不同,给电子体化合物可分为内给电子体和外给电子体。其中,内给电子体在催化剂制备时加入,外给电子体在丙烯聚合时加入。内外给电子体的独立性与互补性影响着聚丙烯催化剂的性能。内给电子体对于载体化的Z-N催化剂发挥了最核心的作用[2-3]:一方面与MgCl2络合,改变了催化剂的催化活性和定向性;另一方面,控制了TiCl4在MgCl2上的数量和分布,防止产生无规活性中心。内给电子体按其结构的不同可分为单酯类、双酯类、二醇酯类、二醚类、二酮类等。
N催化剂是北京化工研究院自主研发的以MgCl2为载体的第四代高效聚丙烯催化剂,通过前期研究可知[4-5],N催化剂中除了可以加入邻苯二甲酸正丁酯(DNBP)、邻苯二甲酸正丁酯(DIBP)等内给电子体外,催化剂本身在制备时原料间也发生反应产生了活性组分,在滴加TiCl4之前体系中产生了PA-DCP-MgCl和DCP-MgCl两种给电子体,这些给电子体的存在影响着N催化剂的颗粒形态及催化性能。因此建立该类给电子体含量的测定方法,对于催化剂机理研究及性能改进具有重要意义。
核磁共振法广泛应用于有机化合物的结构解析和定性分析[6],在化合物纯度定值、含量测定中具有很多优势[7-9]。定量核磁共振法以含氢有机化合物的NMR波谱信号积分面积与氢原子数目成正比为依据,不需引进任何校正因子,不需以每一种被测物的纯品作为参比标准。另外,本文研究对象包含的其中两种给电子体PA-DCP-MgCl和DCP-MgCl,均通过化学反应所得,若采用传统液相色谱的定量方法,无市售标准品可得,给实验增加困难。本文采用核磁共振氢谱内标法,建立了一种快速、专属、简单,用于无对照品的N催化剂中活性组分含量定量的表征方法。
1 实验部分
1.1 仪器与原料
超导核磁共振谱仪:AVANCE 300型,瑞士布鲁克公司。
无水MgCl2、磷酸三丁酯(TBP)、环氧氯丙烷(ECP)、邻苯二甲酸酐(PA);N催化剂:工业级,中国石化股份有限公司催化剂北京奥达分公司提供;无水MgCl2经研磨后使用,TBP和ECP经4A分子筛干燥后使用;对苯二酚(HQ,纯度99.5%),氘代甲醇(Methanol-d4,纯度99.8%),氘代甲苯(Toluene-d8,纯度99.5%),均购于百灵威科技有限公司。
1.2 制备方法
邻苯二甲酸酐溶解液的制备:惰性气体保护下,向反应器中加入0.6 mL氘代甲苯、30 mg无水MgCl2、78.2 μL TBP和 25.5 μL ECP,在56 ℃下反应2.5 h后,加入8.8 mg PA,反应1 h,形成邻苯二甲酸酐溶解液体系。取0.5 mL该反应液置入5 mm的核磁管中,然后放入核磁谱仪探头中进行氢谱测试。
N催化剂按文献[10]的方法制备,添加的内给电子体为DNBP。
1.3 1H NMR表征条件
测定温度:25 ℃,环境湿度:35%,观察频率:300.13 MHz,谱宽:6 172.84 Hz,探头温度298 K,时间域数据点64 k,90°脉冲宽度为12.2 μs,采样时间为5 s,脉冲延迟时间为12 s,累加次数为32。
1.4 实验方法
分别准确称取适量N催化剂和内标物置于直径5 mm核磁共振样品管中,加适量氘代甲醇振荡溶解,制成待测试样溶液。在上述实验条件下调整仪器参数,调谐探头、匀场、采样,得到1H NMR 谱。再进行相位和基线调整,对N催化剂和内标物的定量峰分别进行5次积分,取其平均值,得到积分结果。
以1H NMR内标法,按下式计算体系内活性组分含量[11-12]。
其中:As为被测样品定量峰的积分面积,ns为被测样品定量峰包含的质子数,Ms为被测样品的分子量,Ar为内标物定量峰的积分面积,nr为内标物定量峰包含的质子数,Mr为内标物的分子量,mr为内标物质量,Wr为内标物的质量分数,ms为样品质量。
2 结果与讨论
2.1 N催化剂中活性组分的结构确认及溶剂的选择
N催化剂的制备主要包括4个步骤:(1)无水MgCl2溶解于ECP、TBP和甲苯体系的过程,(2)PA的溶解过程,(3)滴加TiCl4析出活化的MgCl2的过程,(4)酯和钛负载处理、洗涤。
为了对N催化剂制备过程中MgCl2溶解过程和PA溶解过程中产生的新化合物进行化学结构确认,按照“1.2”方法制备了邻苯二甲酸酐溶解液。该溶解液在氘代甲苯中的核磁共振氢谱分辨率不佳(图1B),影响数据的进一步分析。通过对不同氘代溶剂中氢谱的比对分析,将体系中的氘代甲苯减压除去后加入氘代甲醇,得到峰形尖锐、分辨率佳的氢谱图(图1A)。通过谱峰化学位移和积分面积的分析,4组峰dH、cH、bH、aH归属于原料TBP;积分面积比eH∶fH = 4∶1归属于ECP开环后产物DCP-MgCl;积分面积比hH∶iH∶jH∶gH∶hH = 1∶1∶2∶1∶4,归属于PA酸酐环断裂后生成的邻苯二甲酸酯类产物PA-DCP-MgCl。
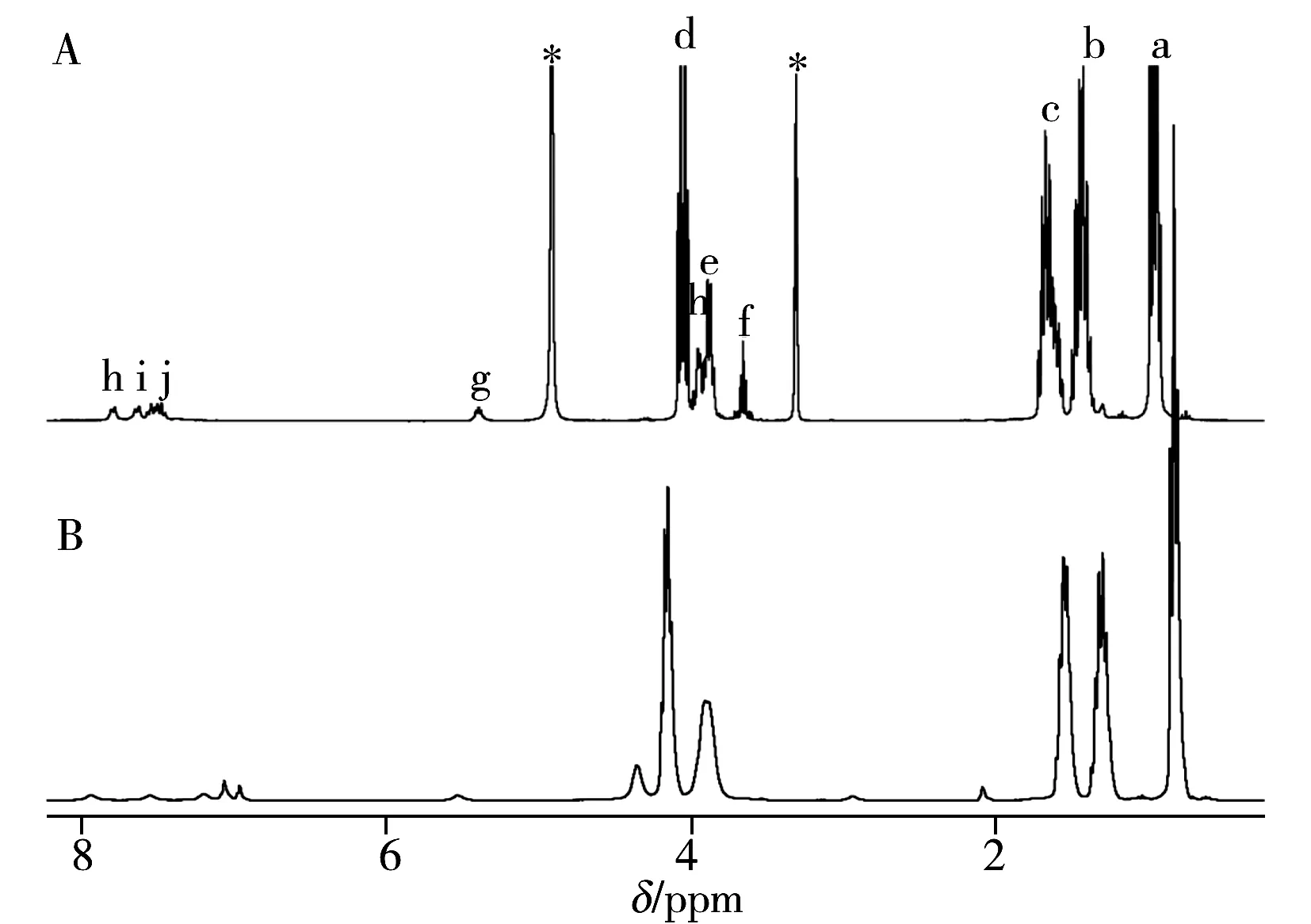
图1 邻苯二甲酸酐溶解液在不同溶剂中的1H NMR谱图Fig.1 1H NMR spectra of PA reaction mixture in different solventsA:methanol-d4;B:toluene-d8

图2 N催化剂核磁共振氢谱内标法中内标物的选择Fig.2 1H NMR spectrum of N catalyst with HQ as internal standard
2.2 内标物及定量峰的选择
内标物质应具有较高的纯度和准确含量,不与样品和溶剂发生化学反应或缔合,能溶于氘代溶剂,含有多个质子,具有易于识别的谱峰等特点,并且内标峰与样品的定量峰能达到基线分离[13]。
根据之前的研究经验[4-5,14],选择对苯二酚为内标物,如图2箭头所示,对苯二酚在δ= 6.6处有一尖锐的单峰,对应于苯环上的4个氢,并与待测物谱峰均无重叠,可作为内标物的定量峰。如图3所示,对于N催化剂中待测的4种活性组分TBP、PA-DCP-TiClx、DCP-TiClx和DNBP,分别选取dH、hH、eH、kH作为它们的定量峰,这几组定量峰峰形尖锐并完全分离,且可与内标物的定量峰明显区分,满足核磁氢谱内标法定量的要求。

图3 N催化剂核磁共振氢谱内标法中定量峰的选择Fig.3 Selection of quantitative peaks in 1H NMR spectrum of N catalyst
2.3 采样时间的选择
核磁谱图是通过采集自由衰减信号(FID)并经傅立叶变换而得到,因此需足够的采样时间以保证自由感应衰减能衰减完全得到分辨率较好的谱图。但过长的采样时间会导致实验时间大大增加,实验中以不同的采集时间采集图谱,结果表明内标物和样品的FID信号均在5 s以内衰减完全,因此将采样时间选定为5 s。
2.4 脉冲延迟时间的选择
核磁定量研究中,延迟时间是最重要的参数之一。一般来讲,延迟时间(D1)为2~5倍于被测物的纵向弛豫时间(T1),延迟时间过短可能造成核的饱和现象,使测定结果偏差较大,而过长的延迟时间会大大增加测试时间[15]。
实验以kH作定量峰为例,按从短(1 s)到长(25 s)分别测定样品的1H NMR,考察被测物定量峰与内标物峰面积之比As/Ar随延迟时间的变化。结果显示,当延迟时间小于12 s时,As/Ar的值随延迟时间的增加而增加,而当延迟时间进一步增加时,As/Ar的值基本不变,因此实验选取D1=12 s。
2.5 定量结果
分别称取5份N催化剂样品,按“1.4”方法,在选定的实验条件下测试1H NMR图谱,并对选取的定量峰分别进行积分,按1H NMR内标法公式计算样品纯度。经计算,N催化剂中4种活性组分TBP、PA-DCP-TiClx、DCP-TiClx和DNBP在催化剂中的摩尔含量分别为0.008、0.013、0.043、0.209 mmol/g。由于N催化剂在制备过程中滴加TiCl4时,除产生一元取代产物外,还可能存在二元、三元或四元取代产物,该体系较复杂,本文将反应后有机产物中的Ti以—TiCl3基团的存在来计算N催化剂中待测的活性组分含量。
称取N催化剂试样和对苯二酚标准样品适量,在同一实验条件下连续做6次1H NMR谱,分别选取其中的kH、dH、eH、hH定量峰与内标定量峰积分面积比进行计算(表1),其相对标准偏差(RSD)分别为0.35%、0.75%、0.41%和0.89%,表明该实验方法的重复性较好。
分别称取5份同一批次的N催化剂试样,按“1.3”方法,在选定的实验条件下做1H NMR图谱,并分别对选取的定量峰和内标峰进行积分,按1H NMR内标法公式计算其中有效组分的含量。结果表明,其中活性组分TBP、PA-DCP-TiCl3、DCP-TiCl3和DNBP的质量分数分别为0.21%、0.58%、1.21%和5.82%,占N催化剂总量的7.82%。在实验中,采用高效液相色谱(HPLC)法以甲醇-水为流动相对该批次的N催化剂样品中的DNBP含量进行测定,测得其含量为6.64%,与核磁内标法测得的含量基本吻合,验证了核磁内标法测定纯度的可靠性。
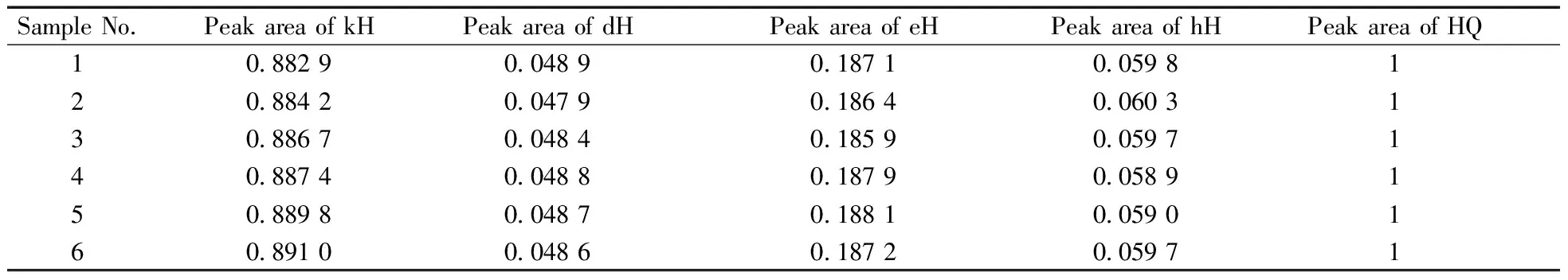
表1 N催化剂与对苯二酚积分面积的测定结果Table 1 The integral area of N-catalyst and HQ
此外,N催化剂制备时,在进行滴加TiCl4析出MgCl2、内给电子体和钛负载处理两个步骤之前的邻苯二甲酸酐溶解液中,各活性组分的含量及浓度也可通过“1.4”方法计算得到。经计算,此时邻苯二甲酸酐溶解液体系中的活性组分为TBP、PA-DCP-MgCl和DCP-MgCl,以HQ为内标物计算它们在体系中的浓度分别为0.408、0.058、0.188 mol/L。由之前的研究结果可知,十分微量的TBP在该体系中发生化学反应,而此时若以TBP为该体系内标物,体系中各活性组分的浓度分别为0.467、0.067、0.167 mol/L,较HQ为内标物时有明显变化,说明在制备N催化剂时,作为助溶剂的TBP在体系中的化学反应量不可忽略。
3 结 论
优选了计算N催化剂体系活性组分的内标物质,以氘代甲醇为溶剂,经谱峰化学位移对比,选择对苯二酚作为内标物质,以核磁内标法测定N催化剂样品中活性组分的含量,溶剂峰和内标定量峰对样品的定量峰无干扰,其结果的重复性和可靠性良好。该方法具有不需标准对照品、快速、高效等特点,在利用核磁技术研究N催化剂中活性组分的化学结构时可同时进行含量测定,为给电子体含量的表征提供了新的途径,从而加深了对N催化剂优异聚合性能的理解,使N催化剂中活性组分的改进和性能的提高有了更多的可能性。
[1] Kashiwa N.J.Polym.Sci.PartA:Polym.Chem.,2004,42 (1): 1-8.
[2] Dong X F,Cui X P,Yang M,Liu B Y.Polym.Mater.Sci.Eng.(董小芳,崔晓鹏,杨敏,刘宾元.高分子材料科学与工程),2017,33 (3): 25-29.
[3] Ling Y T,Xia X Z,Liu Y X.Petrochem.Technol.(凌永泰,夏先知,刘月祥.石油化工),2017,46 (4): 422-426.
[4] Yin J,Zhao M J.Petrochem.Technol.(殷杰,赵梅君.石油化工),2014,44(1): 42-46.
[5] Yin J.Petrochem.Technol.(殷杰.石油化工),2017,46 (1): 124-129.
[6] Deng Z W,Li J,Xu M F,Liu P,Geng Z F.J.Instrum.Anal.(邓志威,李璟,许美凤,刘鹏,耿珠峰.分析测试学报),2012,31(9): 1081-1088.
[7] Deng X J,Li W B,Liu S N,Ding G S.Chin.J.Anal.Lab.(邓小娟,李文斌,刘塞纳,丁国生.分析试验室),2017,36(9): 1032-1035.
[8] Zhang Y J,Liu X P.Chin.J.Magn.Reson.(张友杰,刘小鹏.波谱学杂志),2007,24(3): 289-295.
[9] Zhang W,Xu B.Chem.Anal.Meter.(张伟,徐蓓.化学分析计量),2004,13(6): 39-41.
[10] Zhou Q L,Tan Z,Yan L A,Xu X D,Song W W.Petrochem.Technol.(周奇龙,谭忠,严立安,徐秀东,宋维玮.石油化工),2010,39(9): 997-1000.
[11] Wang Q,Wang M T,Zhang Z Q.J.Instrum.Anal.(王强,汪茂田,张志权.分析测试学报),2003,22(6): 101-103.
[12] Sun J X,Zhang Z X.Chin.J.Pharm.Anal.(孙静霞,张正行.药物分析杂志),2005,25(1): 117-122.
[13] Hu M,Hu C Q,Liu W Y.Chin.J.Anal.Chem.(胡敏,胡昌勤,刘文英.分析化学),2004,32(4): 451-455.
[14] Yin J,Zhao M J,Wang H.Chin.J.Anal.Lab.(殷杰,赵梅君,王红.分析试验室),2014,33(8): 972-974.
[15] Liu K,Wang M C,Xu M,Zhang L H,Zhang G,Chen Z Q.J.Instrum.Anal.(刘可,王民昌,徐敏,张丽涵,张皋,陈智群.分析测试学报),2017,36(3): 414-417.
