5位和7位溴代橙酮衍生物的合成
2018-02-28潘国军
沈 棣,潘国军,苏 超,芦 逵
(天津科技大学生物工程学院,天津 300457)
橙酮(aurones)是植物体内的一种次生代谢产物,属于特殊的黄酮类化合物,普遍存在于植物的根、茎、叶、花、果实中,也有报道称少数海洋生物中也存在橙酮类化合物,橙酮母核结构为 2–(Z)–苯亚甲基–3–(2H)–苯并呋喃酮,由一个环外 C=C 双键连接苯并呋喃和苯环(图 1),是相应的黄酮(flavones)的同分异构体[1].近年来有关橙酮化合物的生物活性和药理作用研究显示其具有抗癌[2]、抗菌[3]、抗寄生虫[4]、抗病毒[5]、抗炎[6]、抗疟等活性.此外,橙酮还可以作为乳腺癌耐药蛋白(ABCG2)抑制剂[7]、P–糖蛋白(Pgp)介导的多药耐药调节剂、乙酰胆碱酯酶抑制剂和脑 B型单胺氧化酶抑制剂[8]等.如图 1所示,Hamiltron(Ⅰ)具有切割 DNA链的作用,化合物Ⅱ对乳腺癌白药蛋白(ABCG2)有抑制作用;Aureusidin(Ⅲ)可作为碘化甲腺氨酸脱腆酶的抑制剂.
天然橙酮化合物多存在于植物当中,但含量低,且因为其结构复杂、作用位点多、溶解性差,导致生物利用率不高,限制了它们的广泛应用.目前,文献报道的有关橙酮化合物的结构修饰,主要集中在C4、C5、C7、C2'、C3'、C4'等位置.通过引入具有不同作用的基团,例如卤素、烷(氧)基、芳基、二甲氨基、氨基、羧基、烯丙基等基团,以期改善其生物活性和提高生物利用率.
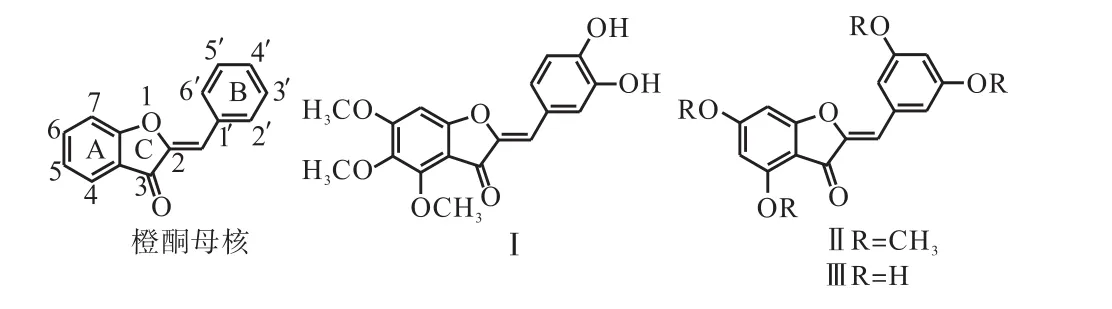
图1 橙酮化合物的结构式Fig. 1 Structures of aurones
宋成荣等[9]在研究橙酮的抑菌活性中发现,B环上有卤素取代的化合物对番茄灰霉病菌的抑制效果较好.黄新炜等[10]对橙酮的抗肿瘤活性研究结果表明,在B环4'位引入卤素和3'位引入吸电子基团有利于增强抗肿瘤活性.张敏等[11]发现在 A环上 5位引入甲基可以显著提高橙酮的除草活性.除上述文献报道外,还发现在查尔酮化合物的3位和5位引入异戊烯基、香叶基可以显著提高其抗肿瘤活性[12].
因此,考虑在橙酮的5位和7位进行结构改造,利用 Suzuki反应作为关键反应引入烷基、芳基等基团,以期改善橙酮的溶解性和生物活性.那么作为Suzuki反应关键底物的5位和7位的卤代橙酮化合物的合成就成了首要解决的问题.此前,本课题组成功地在黄酮化合物的6位和8位实现了选择性碘代和溴代[13–14],并且在使用卤代黄酮化合物进行 Suzuki反应时,溴代黄酮的脱卤副产物明显少于碘代黄酮.与黄酮的选择性卤代研究不同,本研究目的是为了能够简便地合成5位与7位卤代橙酮化合物,为之后对其不同位点进行结构改造得到大量衍生物并进行活性研究提供可能.因此,设计3个橙酮化合物母核,并以此探究5位和7位溴取代橙酮高效简洁的合成方法.
1 材料与方法
1.1 试剂与仪器
二溴海因、N–溴代丁二酰亚胺,化学纯,安耐吉化学;三氯化铝、盐酸、氢氧化钾、间苯三酚、对羟基苯甲醛、3,4–二羟基苯甲醛、3,4,5–三羟基苯甲醛、1,2–二氯乙烷(DCE)、氯乙酰氯、无水硫酸钠、无水碳酸钾、2–碘丙烷;二氯甲烷(DCM),乙醇(EtOH)、石油醚、乙酸乙酯、N,N–二甲基甲酰胺(DMF),分析纯,北京化学试剂公司.
循环水式真空泵,河南省予华仪器有限公司;集热式恒温加热磁力搅拌器,郑州长城科工贸有限公司;低温恒温反应浴,巩义市京华仪器有限公司;Av–400,MHz 型核磁共振仪,瑞士Bruker公司.
1.2 合成路线
设计 5位和 7位溴代橙酮目标产物的合成线路如图2所示.

图2 目标橙酮化合物的合成线路Fig. 2 Synthetic route of target aurones
在对橙酮进行选择性溴代反应时,针对溴化试剂及反应条件作了筛选和优化.起初,采用 NBS作为溴化试剂,在–40,℃时几乎不反应.之后,升温至–20、0、25,℃时底物的转化程度都很低.因而,考虑使用二溴海因(DBDMH)作为溴化试剂,在–40,℃条件下能达到较高的转化率.在室温下反应,底物几乎可以完全转化为5位、7位溴代的目标产物.
以异丙基全保护的金鱼草素为例,在不同条件下的溴化反应(图 3)结果见表 1.在 25,℃条件下,以二溴海因作为溴化试剂,二氯甲烷作为溶剂,就能以较高的收率同时得到 5位、7位溴代橙酮目标化合物,两者的产率分别在53%,和46%,.
1.2.1 化合物11的合成
称取5,g(39.65,mmol)间苯三酚溶于80,mL DCE中,在冰浴条件下,加入 10.57,g(79.30,mmol)无水三氯化铝并搅拌 10,min,然后用恒压滴液漏斗逐滴滴加5.37,g(47.58,mmol)氯乙酰氯,搅拌30,min后升至室温并用碱液作尾气吸收装置,此后将反应体系置于100,℃油浴中加热回流,反应 8,h.冷却至室温后,倒入 0,℃的浓盐酸,搅拌 30,min后抽滤、洗涤,干燥得到4.98,g白色固体化合物11,产率62%,.
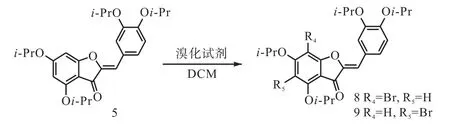
图3 异丙基保护的金鱼草素溴化反应Fig. 3 Bromination of isopropyl protected aureusidin
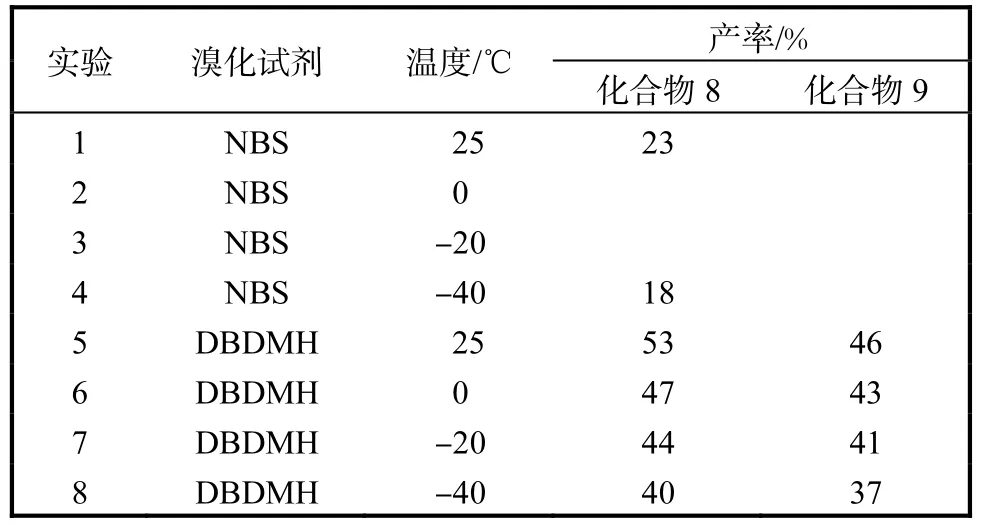
表1 溴化试剂及反应条件的筛选Tab. 1 Screening of bromide reagent and reaction conditions
1.2.2 化合物1—3的合成
称取6.72,g(59.82,mmol)氢氧化钾并配制成质量分数50%,的水溶液,室温搅拌下加入3,mL乙醇以及1.22,g(9.97,mmol)对羟基苯甲醛,最后加入2.02,g(9.97,mmol)化合物 11,体系置于 60,℃油浴中反应5,h,TLC检测反应.待反应完全后,在冰浴下慢慢加入 0,℃的稀盐酸调 pH 至 3~4,抽滤、洗涤,干燥得到2.43,g红色固体化合物1,产率90%,.
用3,4–二羟基苯甲醛和2–羟基苯甲醛替换对羟基苯甲醛,以相同的方法可得到化合物 2和 3,产率分别是 87%,、91%,.
1.2.3 化合物4、5和12的合成
称取2.7,g(10,mmol)化合物1置于100,mL耐压瓶中,加入 10,mL DMF将其溶解,在搅拌状态下加入 5.39,g(39,mmol)无水碳酸钾和 6.63,g (39,mmol)2–碘丙烷(i-PrI),60,℃油浴中反应 24,h.待反应冷却至室温后,滤除体系中固体,用乙酸乙酯洗涤并将滤液倒入 1,mol/L盐酸中.用乙酸乙酯进行萃取,分离得到有机相并将有机相用饱和食盐水洗涤3次,之后用无水硫酸钠进行干燥,旋干有机相,拌样.粗品经过硅胶柱层析(V(石油醚)∶V(乙酸乙酯)=40∶1的混合溶液为展开剂)纯化后,得到 3.09,g黄色固体化合物 4,产率 78%,.
用化合物2以相同的方法可得到化合物5,产率81%,.
用化合物 3以相同的方法可得到化合物 12,产率 84%,.
1.2.4 化合物6、7、8、9、13、14的合成
称取3.97,g(10,mmol)化合物4溶于25,mL干燥的二氯甲烷中,向其中加入 1.72,g(6,mmol)二溴海因,在室温下搅拌反应 30,min.之后将反应体系倒入200,mL冰水中,用二氯甲烷(3×50,mL)萃取,合并有机相并用饱和食盐水萃洗,分离得到有机相用无水硫酸钠干燥.减压旋干有机相,拌样.粗品经过硅胶柱层析(V(石油醚)∶V(乙酸乙酯)=40∶1的混合溶液为展开剂)分离纯化后,同时得到 2.52,g黄色固体化合物6和2.14,g黄色固体化合物7,产率为53%,和45%,.
称取 4.00,g(8.74,mmol)化合物5,以相同的方法可同时得到2.47,g黄色固体化合物8和2.14,g黄色固体化合物9,产率为53%,和46%,.
称取 4.00,g(9.34,mmol)化合物 12,以相同的方法可同时得到2.47,g黄色固体化合物13和2.18,g黄色固体化合物14,产率为52%,和46%,.
1.3 化合物表征数据
化合物 6:1,H NMR(400,MHz,CDCl3)δ 7.87(d,J=8.8,Hz,2H),6.96(d,J=9.2,Hz,2H),6.75(s,1H),6.20(s,1H),4.88~4.82(m,1H),4.71~4.60(m,2H),1.45(d,J=6.0,Hz,6H),1.43(d,J=6.0,Hz,6H),1.37(d,J=6.0,Hz,6H).13,C,NMR(101,MHz,CDCl3)δ 180.29,164.74,162.46,159.26,157.28,146.43,133.28,124.76,116.07,111.89,107.46,96.48,86.88,73.44,72.86,69.99,58.49,22.01,18.45. 熔点 138.6,℃.LRMS m/e C24,H27BrO5[M+H]+理论值475.1,实测值475.1.
化合物 7:1H NMR(400,MHz,CDCl3)δ 7.81(d,J=8.8,Hz,2H),6.94(d,J=8.8,Hz,2H),6.74(s,1H),6.54(s,1H),5.25~5.19(m,1H),4.73~4.60(m,2H),1.48(d,J=6.0Hz,6H),1.41(d,J=6.0,Hz,6H),1.37(d,J=6.0Hz,6H).13,C,NMR(101,MHz,CDCl3)δ 179.69,167.34,162.42,159.27,154.88,146.32,133.07,124.74,115.97,111.94,108.18,102.70,92.21,79.09,72.86,70.01,22.65,21.99,21.91. 熔点 107.1,℃.LRMS m/e C24H27BrO5[M+H]+理论值475.1,实测值475.1.
化合物 8:1H NMR(400,MHz,CDCl3)δ 7.73(d,J=2.0,Hz,1H),7.37(dd,J=8.4,2.0,Hz,1H),6.95(d,J=8.4,Hz,1H),6.72(s,1H),6.20(s,1H),4.88~4.82(m,1H),4.69~4.54(m,3H),1.44(t,J=6.0,Hz,12H),1.41(d,J=6.0,Hz,6H),1.37(d,J=6.0,Hz,6H).13,C NMR(101,MHz,CDCl3)δ 180.21,164.67,162.48,157.32,150.71,149.07,146.54,126.09,125.79,119.47,116.93,112.12,107.44,96.50,86.82,73.43,72.90,72.11,72.06,22.37,22.20,22.00. 熔点172.6,℃.LRMS m/e C27H33BrO6[M+H]+理论值 533.1,实测值533.1.
化合物 9:1H NMR(400,MHz,CDCl3)δ 7.46(d,J=2.0,Hz,1H),7.45(d,J=2.0,Hz,1H),6.95(d,J=9.0,Hz,1H),6.70(s,1H),6.52(s,1H),5.25~5.18(m,1H),4.74~4.67(m,1H),4.62~4.56(m,1H),4.54~4.48(m,1H),1.48(d,J=6.0,Hz,6H),1.41(d,J=6.0,Hz,6H),1.38(d,J=6.0,Hz,6H),1.37(d,J=6.0,Hz,6H).13C NMR(101,MHz,CDCl3)δ 179.65,167.35,162.45,154.89,151.24,148.63,146.46,126.33,125.58,121.71,116.44,112.04,108.16,102.78,92.25,79.07,72.96,72.88,71.72,22.63,22.27,22.15,21.90. 熔点 169.5,℃.LRMS m/e C27H33BrO6[M+H]+论值533.1,实测值533.1.
化合物 13:1H NMR(400,MHz,CDCl3)δ 8.35(dd,J=7.9,1.4,Hz,1H),7.38(s,1H),7.34~7.30(m,1H),7.06(t,J=15.2,Hz,1H),6.92(d,J=8.4,Hz,1H),6.21(s,1H),4.92~4.86(m,,H),4.71~4.58(m,2H),1.45(d,J=6.0,Hz,6H),1.42(d,J=6.0,Hz,6H),1.38(d,J=6.4,Hz,6H).13,C,NMR(101,MHz,CDCl3)δ 180.51,164.96,162.59,157.42,157.31,147.54,132.20,131.02,122.64,121.00,113.65,107.58,106.26,97.05,87.13,73.83,72.92,71.23,22.16,22.14,22.07. 熔点 125.2,℃.LRMS m/e C24,H27BrO5[M+H]+理论值475.1,实测值475.1.
化合物 14:1H NMR(400,MHz,CDCl3)δ 8.22(dd,J=7.8,1.4,Hz,1H),7.36(s,1H),7.32~7.28(m,1H),7.00(t,J=15.2,Hz,1H),6.91(d,J=8.4,Hz,1H),6.53(s,1H),5.28~5.21(m,1H),4.71~4.58(m,2H),1.46(d,J=6.0,Hz,6H),1.41(d,J=6.0,Hz,6H),1.37(d,J=6.0,Hz,6H).13C,NMR(101,MHz,CDCl3)δ 179.90,167.55,162.57,157.24,154.98,147.45,131.91,131.12,122.42,120.57,113.42,108.16,106.39,102.73,92.31,79.13,72.94,70.95,22.76,22.14,21.97. 熔点 124.5,℃.LRMS m/e C24H27BrO5[M+H]+理论值475.1,实测值475.1.
2 结果与讨论
2.1 橙酮母核的合成
传统的合成橙酮的方法一般有两种:第一,以3(2H)–苯并呋喃酮为关键中间体;第二,通过 2′–羟基查耳酮的氧化环化.然而第一种方法中 3(2H)–苯并呋喃酮的合成实验条件比较苛刻,且对一些底物的反应产率较低;第二种方法对底物结构有限制,且反应步骤多,较繁琐.
以化合物2金鱼草素为例,本实验中橙酮母核的合成是通过化合物 11与 3,4–二羟基苯甲醛发生缩合、关环、消除反应得到,反应操作简单,收率也较高.具体反应机理如图 4所示,首先化合物 11在碱性条件下,失去羰基α 氢形成α 碳负离子进攻醛的碳正离子,氯离去,形成环氧中间体;然而由于环氧结构的不稳定,氧拉电子导致两个位置的碳呈正电性,此外该结构中的羰基同样具有吸电子作用,使得与之相邻的碳更显正电,因而苯环上的氧更容易进攻羰基的α 碳;另一方面,从空间位阻的角度考虑,在碱催化的条件下,氧更容易进攻空间位阻小的碳.所以,综合电子效应和空间位阻两个因素来分析,在环氧开环这一步中,氧更容易进攻羰基的α 碳,从而形成的是闭合的五元环结构,而不是六元环.最后在碱性加热的条件下,羟基消除生成环外双键,得到最终的橙酮化合物——金鱼草素.
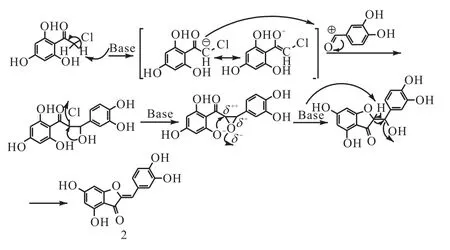
图4 橙酮母核合成反应机理Fig. 4 Reaction mechanism of the aurone
在处理该反应的过程中,由于金鱼草素上的酚羟基具有酸性,所以在氢氧化钾的作用下,金鱼草素是以水溶性很好的钾盐的状态存在,然而加入盐酸后使其变回酚羟基,溶解度降低,因此产物可以从溶液中析出,得到金鱼草素的粗产品.接下来可以利用金鱼草素不溶于二氯甲烷,而部分杂质溶于二氯甲烷的性质,将干燥后的粗品用二氯甲烷溶解、洗涤、抽滤,进行纯化可得纯度较高的金鱼草素.
2.2 橙酮的溴代反应
在以二溴海因作为溴化试剂进行橙酮的选择性溴代时,发现其选择性较差.一般以 53%,和 45%,左右的产率分别得到 7位的溴代产物和 5位的溴代产物.分析其原因可能是:一方面橙酮与黄酮的结构相比,橙酮的中间是五元环而黄酮的中间是六元环,橙酮3位上的羰基对4位的异丙基造成的挤压小,因而在 5位拥有了更大的空间,因此 Br原子更容易进攻5位;另一方面由于橙酮的双键在环外,因此 C环上的羰基和氧对于 A环的电子云影响也不如黄酮显著,因而5位与7位的亲核性也几乎没太大差别.所以综合分析两种因素可以得出结论:橙酮化合物7位的溴代选择性略高于5位,但两者没有显著区别.
因此,可以近乎相当的产率同时得到5位和7位溴代的橙酮化合物.
3 结 语
本文首先合成了3个橙酮化合物母核,再以它们为重要中间体,通过异丙基保护后,采用二溴海因作为溴代试剂,以中等收率同时获得 5位和 7位的 Br取代橙酮化合物,产率分别在 46%,和 53%,左右,总收率在 99%,左右.该类化合物的成功合成为进一步对橙酮化合物的 5位和 7位进行结构修饰和改造奠定了重要的基础.
[1] Carrasco M P,Newton A S,Gonçalves L,et al. Probing the aurone scaffold against Plasmodium falciparum:Design,synthesis and antimalarial activity[J]. European Journal of Medicinal Chemistry,2014,80:523-534.
[2] Lee C Y,Chew E H,Go M L. Functionalized aurones as inducers of NAD(P)H:Quinone oxidoreductase 1 that activate AhR/XRE and Nrf2/ARE signaling pathways:Synthesis,evaluation and SAR[J]. European Journal of Medicinal Chemistry,2010,45:2957–2971.
[3] Okombi S,Rival D,Bonnet S,et al. Discovery of benzylidenebenzofuran-3(2H)-one(aurones)as inhibitors of tyrosinase derived from human melanocytes[J]. Journal of Medicinal Chemistry,2006,49(1):329-333.
[4] Kayser O,Kiderlen A F,Folkens U,et al. In vitro leishmanicidal activity of aurones[J]. Planta Med,1999,65(4):316-319.
[5] Haudecoeur R,Ahmed-Belkacem A,Yi W,et al. Discovery of naturally occurring aurones that are potent allosteric inhibitors of Hepatitis C virus RNA-dependent RNA polymerase[J]. Journal of Medicinal Chemistry,2011,54(15):5395-5402.
[6] Shin S Y,Shin M C,Shin J S,et al. Synthesis of aurones and their inhibitory effects on nitric oxide and PGE(2)productions in LPS-induced RAW264.7 cells[J]. Bioorganic & Medicinal Chemistry Letters,2011,21(15):4520-4523.
[7] Sim H M,Loh K Y,Yeo W K,et al. Aurones as modulators of ABCG2 and ABCB1:Synthesis and structureactivity relationships[J]. Chemmedchem,2011,6(4):713-724.
[8] Sheng R,Xu Y,Hu C,et al. Design,synthesis and AChE inhibitory activity of indanone and aurone derivatives[J]. European Journal of Medicinal Chemistry,2009,44(1):7-17.
[9] 宋成荣. 橙酮类似物的合成及抑菌活性研究[D]. 杨凌:西北农林科技大学,2012.
[10] 黄新炜,王征,陈巧利,等. 橙酮衍生物的合成及初步抗肿瘤活性研究[J]. 有机化学,2013(12):2565-2571.
[11] 张敏. 苯并[c]菲啶和橙酮类化合物的设计、合成与生物活性研究[D]. 天津:南开大学,2009.
[12] Wang H M,Zhang L,Liu J,et al. Synthesis and anticancer activity evaluation of novel prenylated and geranylated chalcone natural products and their analogs[J]. European Journal of Medicinal Chemistry,2015,92:439-448.
[13] Lu K,Chu J,Wang H M,et al. Regioselective iodination of flavonoids by N-iodosuccinimide under neutral conditions[J]. Tetrahedron Letter,2013,54:6345–6348.
[14] Pan G J,Yang K,Ma Y T,et al. Synthesis of 6- or 8-bromo flavonoids by regioselective mono-bromination and deprotection protocol from flavonoid alkyl ethers[J]. Bulletin of the Korean Chemical Society,2015,36:1460-1466.
