蛛网膜下腔出血大鼠海马区A-β蛋白表达 与认知功能障碍的关联研究
2018-02-27郝璞珩
何 娟,吴 琼,李 娜,郝璞珩
(1. 西安交通大学第一附属医院康复医学科,陕西西安 710061;2. 西安交通大学生物医学信息工程教育部 重点实验室,生命科学与技术学院,陕西西安 710049;3. 西安医学院第一附属医院神经外科,陕西西安 710077)
蛛网膜下腔出血(subarachnoid hemorrhage, SAH)通常是由于脑内动脉瘤破裂导致血液流入蛛网膜下腔而引起的病变,在人类具有高致残率和高死亡率的特点。SAH的病理过程包括灌注减少、微血栓形成、大脑水肿以及迟发性脑缺血,继而引起不同程度的感觉、运动、认知等方面功能损伤[1-2]。A-β是淀粉样前体蛋白(amyloid precursor protein, APP)在溶酶体、高尔基体、内质网的异常代谢产物。A-β表达增加,沉积在新皮质、杏仁核及海马等部位,可以导致神经元损伤及认知功能减退[3-4]。近年来A-β对神经系统损伤的研究颇多,但大多集中于阿尔兹海默病的认知功能的研究,对于SAH后认知功能障碍与A-β的关系研究尚少。
本实验通过前瞻性研究, 对蛛网膜下腔出血大鼠海马组织A-β蛋白表达及认知功能的动态变化进行观察,探讨其与病情及预后的关系。
1 材料与方法
1.1 主要仪器和试剂 江苏医疗器械厂生产的解剖剪、眼科剪、刀柄刀片、显微有齿镊及无齿镊、三角针及小圆针;Olympus BX51 荧光显微镜(日本)。
A-β1-40单克隆抗体(美国Bioword Technology公司);生物素化山羊抗小鼠抗体、封闭用正常羊血清、DAB显色试剂盒、辣根酶标记链酶卵白素(北京中杉金桥生物技术有限公司)。
1.2 实验动物 成年健康雄性SD(Sprague-Dawley)大鼠48只,体质量250~280 g,购自西安交通大学医学部实验动物中心。于实验动物中心SPF环境下饲养,统一喂养配制的鼠粮和水,饲养1周后进行实验。随机分为假手术组和模型组,每组又分为3、7、14、28 d 4个亚组。
1.3 大鼠SAH模型的建立 参照Bederson法[5],采用刺破颈内动脉法建立SD大鼠SAH模型。采用3-0单股的直径0.2 μm、长50 mm尼龙线将头端钝化,于距头端18.5 mm处作标记,750 mL/L乙醇清洁后放置在肝素化的生理盐水中备用。SD大鼠称重后用100 g/L水合氯醛(300 mg/kg)腹腔注射麻醉后取仰卧位固定,取腹侧颈部正中切口,钝性分离软组织后暴露右侧颈总动脉和颈动脉分叉处,分离颈内动脉和颈外动脉,结扎并切断颈内动脉和颈外动脉之间的吻合支,然后将颈外动脉结扎并切断,将其尾端拉直与颈内动脉成一条直线,用直径为0.2 μm钝化后的尼龙线从颈外动脉插入颈内动脉颅内段,插入深度约为18.5 mm,使线插进大脑中动脉开口处,感觉到阻力时再快速前进3 mm刺破血管,制造SAH模型。15 s后迅速将尼龙线拔出,并扎紧颈外动脉断端,缝合皮肤切口。假手术组大鼠除了不刺破血管壁外,其余操作均与模型组一致。2组大鼠均于麻醉苏醒后继续于实验动物中心SPF实验室中饲养,正常喂食和水。
1.4 Morris水迷宫实验流程 使用自制的Morris水迷宫装置,主要由盛水的圆形水池(直径140 cm,高60 cm)和一个可移动位置的黑色平台(直径10 cm,高35 cm)组成。水池壁上标有东、南、西、北4个注水点,4个注水点将水池划分为4个象限,注水深度为40 cm,使用少量墨汁将水染黑。本实验选取第1象限中央放置移动平台。分别于术后3、7、14、28 d进行水迷宫检测。每只大鼠分别于4个象限的同一位置放入水池,分别计算4个象限到达平台的时间,计算4个象限到达平台的时间均数为逃避潜伏期时间,撤离平台后计算大鼠在原象限滞留的时间之和为象限滞留时间。
1.5 取材行A-β免疫组化染色 用100 g/L水合氯醛(300 mg/kg)腹腔麻醉,继而开胸,将针头由左心室插入主动脉,使用止血钳固定,同时剪开右心耳。生理盐水200 mL进行灌注,再使用40 g/L多聚甲醛200 mL进行灌注。然后快速取下全脑组织,将全脑组织用40 g/L多聚甲醛固定24 h后,取视交叉后0.54~6.54 mm区域制作常规石蜡包埋切片,行A-β(滴度1∶100)免疫组化染色,贴片封蜡。

2 结 果
2.1 2组大鼠逃避潜伏期的比较 手术前所有大鼠神经功能均正常,模型组与假手术组逃避潜伏期时间无明显差异(P>0.05)。造模后相应时间点,模型组比假手术组逃避潜伏期时间明显延长(P<0.05),象限滞留时间明显减少(P<0.05,表1)。
2.2 2组A-β蛋白表达的比较 免疫组化染色结果显示,A-β标记的神经元,以细胞核含有棕黄色的颗粒或斑片的为阳性。模型组在造模后3、7、14 d的A-β阳性细胞计数持续升高,14 d达到峰值,28 d的A-β阳性细胞计数较前下降。在7、14、28 d时,模型组与假手术组的A-β阳性细胞计数具有统计学差异(P<0.05,表2、图1)。
表1 2组大鼠逃避潜伏期时间和象限滞留时间的比较

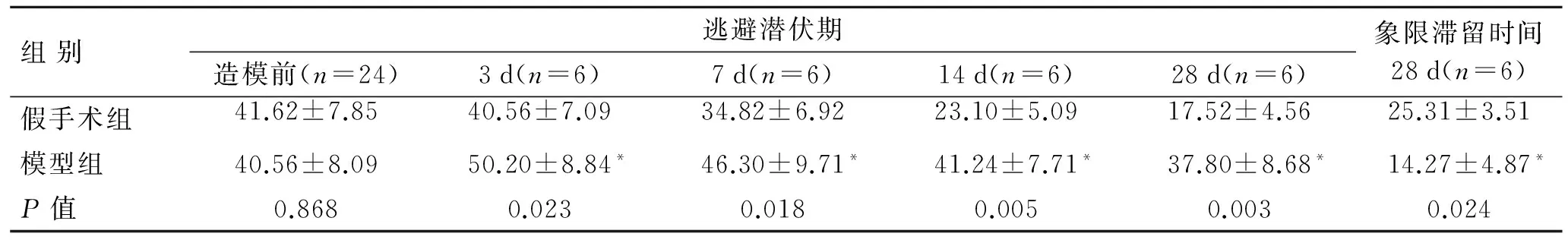
与相应时间点假手术组比较,*P<0.05。
表2 2组大鼠A-β阳性细胞计数的统计比较

与相应时间点假手术组比较,*P<0.05。
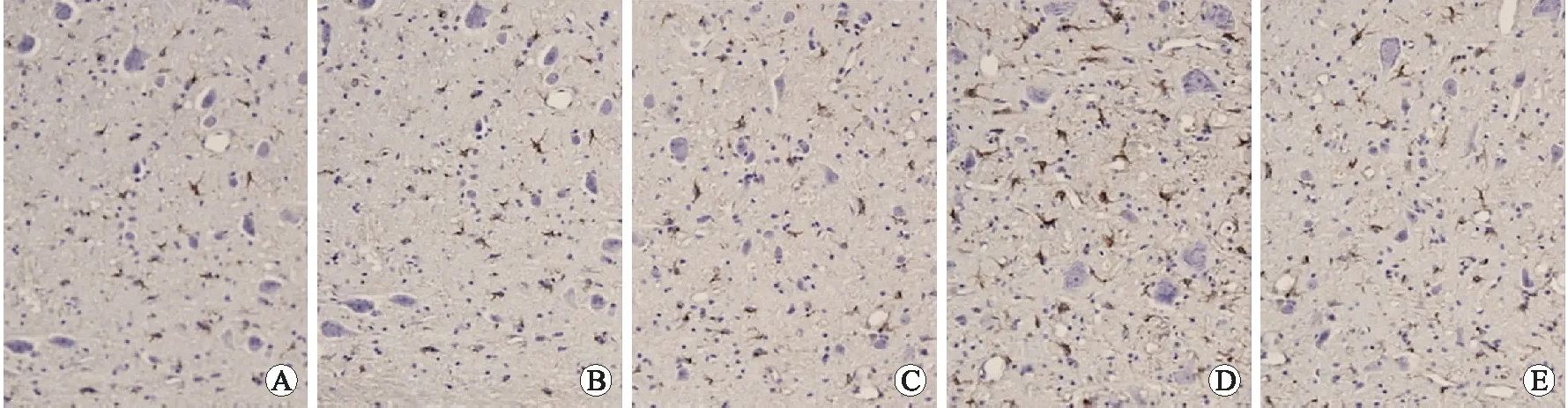
图1 各组大鼠海马部位A-β阳性表达
Fig.1 Positive expression of A-β peptide in the hippocampus of rats (×400)
A:假手术组;B、C、D、E:分别是模型组3、7、14、28 d时间点亚组。
3 讨 论
SAH的病理过程包括微循环灌注减少、大脑水肿、血栓形成以及迟发性脑缺血等,而脑血管痉挛是SAH的主要并发症,使得发病后2周患者的死亡率增加1.5~2倍[6]。血管痉挛造成颅内动脉血管收缩,导致血流量绝对减少,继而引起脑组织灌注不足,从而产生严重的缺血缺氧性神经系统功能障碍,甚至会导致缺血性脑组织坏死[7]。本实验选取SD大鼠,在SAH造模后对神经系统功能障碍中的认知功能改变及相应组织化学指标A-β变化进行观察,探讨与病情变化及预后的关系。
临床痴呆的患者,在患病早期一般先出现遗忘型轻型认知功能障碍,表现为记忆力损害[8]。运用水迷宫来评定大鼠认知功能中的记忆功能是比较恰当的。水迷宫试验中发现,造模前2组大鼠的逃避潜伏期统计无明显差异,造模后模型组大鼠寻找平台的轨迹表现为随机性,表明其学习记忆能力有所下降;而假手术组大鼠在经过几次训练后,大鼠的记忆功能得到强化,逐渐转为直线到达。模型组大鼠水迷宫潜伏期时间较假手术组明显延长,寻找平台错误的次数较假手术组明显增多,相应时间点均具有统计学差异。同一时间点空间探索的时间,模型组和假手术组大鼠分别在4个象限内寻找到平台时间无明显差异,但模型组比假手术组寻找平台的平均时间明显延长。撤离平台后大鼠的象限滞留时间模型组较假手术组明显减少。由此可见,模型组的大鼠出现明显空间学习记忆功能下降。
近年来,A-β在神经变性疾病中的作用备受关注,其在阿尔茨海默病中的研究较为深入,而在SAH所导致的生理、病理过程中的作用却少有研究。A-β是淀粉样前体蛋白,在正常的老年人脑组织中也存在,但其含量很低,是神经元和胶质细胞正常代谢的产物。正常生理条件下,A-β的产生、降解和清除是一个动态平衡过程,当发生基因突变或代谢因素、环境因素改变等作用,引起A-β表达增加、细胞外酶降解水平降低或转运至脑外机制受阻,进而在脑组织中沉积增加,导致神经元损伤进而引起认知功能减退。研究显示A-β具有神经毒性作用,促进细胞膜磷脂超氧化,并抑制磷酸化过程,最终导致细胞损伤和死亡[3-4]。
但认知功能障碍与中枢神经系统哪些部位受损密切相关呢?近年研究显示,海马接受大脑皮质传递的信息并进行选择性储存及记忆,而海马特别是海马的中区是对缺血性损伤最为敏感的脑区之一。目前研究认为,海马这种选择性缺血易损性与学习、记忆功能障碍密切相关,可能是痴呆的病理生理基础之一。而SAH会继发明显的血管痉挛,继而引起神经元损伤,是痴呆的重要病理形态学基础,海马区锥体细胞也经历了缺血、水肿等改变,逐渐发展为基质疏松、微空泡形成、损伤脱失。因此,本实验选择海马作为观察区。目前的研究已经证实,A-β是大脑皮质老年斑的核心成分,也是认知功能障碍病理改变的特征性成分。在脑内产生的A-β主要通过细胞内吞、细胞外酶降解和转运出脑等途径得以清除,一般不会对机体产生影响,但其堆积过多时这种平衡被打破,继而沉积在杏仁核、海马及新皮质等区域[8-10]。
本实验显示,模型组海马局部神经元出现神经元纤维走行紊乱且大片缺失,并出现增粗、肿胀、断裂等损害表现,且A-β标记的神经元较假手术组明显增多,说明海马区的A-β沉积是病理发生的早期关键事件,引发大鼠海马区神经元退行性变的恶性循环及级联放大的病理生理系统。本实验模型组海马区神经元A-β表达明显增多,结合其逃避潜伏期延长,我们推测SAH大鼠继发血管痉挛后神经细胞损伤继而认知功能下降可能与以下机制有关[9-11]:①神经元对各种伤害性刺激如缺血缺氧、炎性反应、自由基损伤、氧化应激等的敏感性增强,从而导致神经元凋亡及坏死;②缺血缺氧损伤后,血脑屏障受到破坏,引起A-β基因被激活而过表达,同时细胞内吞、细胞外酶降解和转运出脑等清除功能下降,A-β在脑内不断蓄积,经一系列的关联反应,最终导致神经元损伤及认知功能障碍。
由于A-β具有不同亚型,且实验的不同浓度等因素对结果有一定影响,并且神经元相互作用涉及到多条路径,所以其具体机制还需要进一步的研究。
[1] MAYOR S. Earlier diagnosis is needed to reduce deaths and disability from aneurysmal subarachnoid haemorrhage[J]. BMJ, 2013, 347:6925.
[2] MOHAMED S, RIVA R, CONTIN M. Simple and validated UHPLC-MS/MS analysis of nimodipine in plasma and cerebrospinal fluid of patients with subarachnoid haemorrhage[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2016, 1028:94-99.
[3] BOERBOOM W, HEIJENBROK-KAL MH, KHAJEH L, et al. Differences in cognitive and emotional outcomes between patients with perimesencephalic and aneurysmal subarachnoid haemorrhage[J]. J Rehabil Med, 2014, 46(1):28-32.
[4] ZHU B, JIANG L, HUANG T, et al. ER-associated degradation regulates Alzheimer’s amyloid pathology and memory function by modulating γ-secretase activity[J]. Nat Commun, 2017, 8(1):1472-1488.
[5] OTITE F, MINK S, TAN CO, et al. Impaired cerebral autoregulation is associated with vasospasm and delayed cerebral ischemia in subarachnoid hemorrhage[J]. Stroke, 2014, 45(3):677-682.
[6] NYBERG C, KARLSSON T, HILLERED L, et al.Metabolic pattern of the acute phase of subarachnoid hemorrhage in a novel porcine model: Studies with cerebral microdialysis with high temporal resolution[J]. PLoS One, 2014, 9(6):e99904.
[7] ROWLAND MJ, HADJIPAVLOU G, KELLY M et al. Delayed cerebral ischaemia after subarachnoid haemorrhage: Looking beyond vasospasm[J]. Br J Anaesth, 2012,109(3):315-329.
[8] MAMM AP, SCODELLER, HUSSAIN S. Identification of a peptide recognizing cerebrovascular changes in mouse models of Alzheimer’s disease[J]. Nat Commun, 2017, 8(1):1403-1414.
[9] HANSON JE, PARE JF, DENG L, et al. Altered Glun2B NMDA receptor function and synaptic plasticity during early pathology in the PS2APP mouse model of Alzheimer’s disease[J]. Neurobiol, 2015, 74:254-262.
[10] MOTA SI, FERREIRA IL, REGO AC. Dysfunctional synapse in Alzheimer’s disease— A focus on NMDA receptor[J]. Neuropharmacology, 2014, 76:16-26.
[11] VAN BEEK AH, CLAASSEN JA. The cerebrovascular role of the cholinergic neural system in Alzheimer’s disease[J]. Behav Brain Res, 2011, 221(2):37-42.
