硒蛋白S截短和全长重组质粒的构建与表达
2018-02-27黄芙萌马芳霞田李芳
黄芙萌,赵 莉,马芳霞,陈 钊,王 莉,田李芳
(西安交通大学第二附属医院肾病内科,陕西西安 710004)
硒(selenium, Se)是多种生物体内不可缺少的微量元素。硒以硒蛋白形式在体内发挥其生物学功能[1]。硒蛋白的特征为蛋白中都有硒代半胱氨酸(selenocysteine, Sec),人体内共有25种硒蛋白基因,编码超过30种硒蛋白[2]。硒蛋白S(SelS)是硒蛋白家族的主要成员,具有抗氧化损伤,参与内质网应激及炎症反应,参与精子发育过程等多方面的生物学功能[3]。人类SelS基因位于15号染色体长臂上,其mRNA有2个转录变体,分别由1 250和1 292个碱基组成,它们均能编码由189个氨基酸残基组成的 SelS,其中第188位为Sec残基,是肽链中倒数第2位的氨基酸[4]。
Se元素以Sec的形式插入硒蛋白,此过程需要一个能将mRNA中的UGA密码子翻译成Sec的特殊机制,在硒蛋白mRNA 3′端非翻译区域(3′TUR)中存在一个茎环结构硒代半胱氨酸插入序列(selenocysteine insertion sequence, SECIS),该结构在翻译过程中能使丝氨酰-tRNA上的反密码子ACU与密码子UGA交错配对[5]。在Sec特异性延长因子、SECIS结合蛋白2(SECIS binding protein 2, SBP2)、核糖体蛋白L30、RNA结合蛋白、可溶性肝脏抗原蛋白和硒磷酸合酶1的辅助下,将Sec-tRNASec上携带的Sec插入到硒蛋白肽链中[6]。
硒蛋白作为机体内硒的最主要代谢形式,在硒缺乏与否的情况下存在两种剪接方式:一种是将UGA翻译为Sec插入硒蛋白肽链中,另一种是UGA作为终止密码子终止硒蛋白肽链的翻译。后者发生在细胞环境中缺乏合成原料硒时,硒蛋白的合成受到限制,而选择将UGA作为终止密码子终止硒蛋白肽链的翻译,不再插入Sec,形成羧基端缺失-Sec-Gly、无酶活性、截短的硒蛋白[7]。
本研究将模拟硒缺乏环境,构建能翻译出截短 SelS的重组质粒,同时构建其具有正常生物活性的全长 SelS的重组质粒,用于后续实验,以期观察硒蛋白S截短体和完整形式在细胞系或动物体内高表达的生物学效应。
1 材料与方法
1.1 材料 本实验在西安交通大学生物化学与分子生物学系实验室完成。pEGFP-C1真核表达载体、人宫颈癌细胞Hela细胞系及DH5α大肠杆菌菌种由本实验室留存。高保真Taq酶及DNA marker购于日本TaKaRa公司,PCR primers由北京三博志远公司合成,RevertAidTMFirst Strand cDNA Synthesis Kit、ApaⅠ酶及XhoⅠ酶购于加拿大Fermentas公司,Gel Extraction System B VER2.0试剂盒购自北京博大泰克生物基因技术有限责任公司,T4 DNA连接酶购于美国Sigma公司,TIANprep Mini Plasmid Kit试剂盒购自北京天根生物技术公司,胰蛋白胨及酵母提取物购于英国Oxoid公司,DMEM高糖培养基购自赛默飞世尔生物试剂,胎牛血清及胰蛋白酶购自美国HyClone公司,Lipofectamine 2000及Trizol®Reagent购自美国Invitrogen公司,E.Z.N.A. Fastfilter Endo-Free Plasmia Midi Kit购于OMEGA生物技术公司,6孔板购于美国Corning公司,倒置相差显微镜及正置荧光显微镜购于日本Olympus公司,流式细胞仪(FACS)购于美国Guava Technologies公司。
1.2 SelS基因PCR引物的设计与合成 截短的SelS用trun-S表示。trun-S中PCR的目的基因片段为CDS部分,不包括5′UTR或者3′TUR;全长SelS中PCR的目的基因片段为CDS+3′TUR部分,不包括5′UTR,3′TUR中含有SECIS序列。设计引物时,先在GenBank上查找出基因序列GenBank No.NM_018445.4,然后利用PRIMER-v5软件进行设计,引物设计好之后在GenBank数据库中利用BLAST 进行查询,确定与其他基因无同源性,为特异性序列。设计时引物的上下游分别引入与载体相应的内切酶位点,上游用核酸内切酶XhoⅠ,其识别序列为CTCGAG,下游用核酸内切酶ApaⅠ,其识别序列为GGGCCC。PCR引物由北京三博远志生物技术有限责任公司合成(表1)。
表1 硒蛋白PCR引物信息
Tab.1 Primer information of selenoproteins

基因引物序列(5'~3')扩增长度(bp)退火温度(℃)SelS正义链CCGCTCGAGATGGAACGCCAAGAGG反义链GCGGGCCCTTAATATACAGAAACAAACCCCATC103460trun-SelS正义链CCGCTCGAGATGGAACGCCAAGAGGAGTC反义链GCGGGCCCTTAGCCTCATCCGCCAG58759
1.3 表达载体的选择 在表达载体pEGFP-C1的多克隆酶切位点上,选择与SelS基因两端对应的核酸内切酶位点XhoⅠ及ApaⅠ,两酶切位点之间的序列将被切除,插入本研究的目的SelS基因片段(图1)。
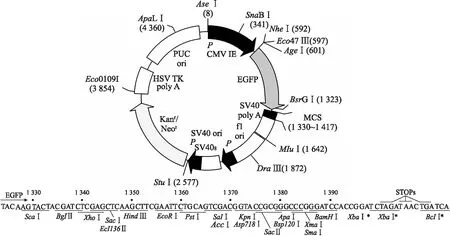
图1 pEGFP-C1载体结构和多克隆位点示意图
Fig.1 Schematic diagram of pEGFP-C1and its multiple cloning site
1.4 重组质粒的构建 建立PCR反应体系:5×Taq酶buffer 10 μL,正义链引物P1 1 μL,反义链引物P2 1 μL,模板0.5 μL,高保真Taq酶0.5 μL,dNTP 4 μL,ddH2O 33 μL。PCR反应条件为:95 ℃ 10 min,95 ℃ 45 s,退火温度45 s,72 ℃ 1 min,34个循环后,72 ℃ 10 min。10 g/L琼脂糖凝胶电泳确定DNA大小为目的条带并且无其他杂带。
用Gel Extraction System B VER2.0试剂盒对PCR产物进行纯化回收。建立50 μL双酶切目的DNA片段的反应体系:XhoⅠ 2 μL,ApaⅠ 2 μL,10×Tango buffer 5 μL,DNA片段20 μL,H2O 21 μL;建立40 μL双酶切载体的反应体系:XhoⅠ 1.6 μL,ApaⅠ 1.6 μL,10×Tango buffer 4 μL,DNA片段10 μL,H2O 22.8 μL。分别于37 ℃水浴6 h。将双酶切获取的目的DNA片段和载体按照上述的胶回收方法回收。建立20 μL连接反应体系:DNA片段8 μL,载体4 μL,T4 DNA连接酶1 μL,10×连接buffer 2 μL,H2O 5 μL,于16 ℃水浴连接过夜。
1.5 转化及克隆的鉴定 常规方法制备感受态DH5α大肠杆菌,将5 μL连接产物加入200 μL感受态细胞过夜培养。第2天用灭菌牙签挑起LB平板上长出的单克隆菌落,移至5 mL LB培养液(含卡纳霉素)中,37 ℃摇菌过夜。存菌种1 mL。用TIANprep Mini Plasmid Kit试剂盒提取扩增好的质粒,进一步做目的硒蛋白基因的PCR扩增,再用琼脂糖凝胶电泳初步鉴定重组质粒中目的硒蛋白基因的转入情况。将已存的500 μL菌液送去公司测序。将测序返回的结果输入NCBI的BLAST软件进行比对。
1.6 转染表达及效率检测 DMEM高糖培养基加100 mL/L胎牛血清常规培养人宫颈癌Hela细胞。转染时将实验设计为4组,分别为Mock组、空载体对照组、trun-SelS组、SelS组,每组设3个平行孔,整个实验共计12孔。6孔板中每孔接种2×105个悬浮在2 mL培养液中的细胞,接种24 h后细胞完全贴壁,当细胞融合度约80%时,每孔加入2 mL不含胎牛血清的DMEM高糖培养基;取无菌EP管1个,分别加入脂质体5 μL和质粒DNA 2 μg,加入无血清培养基,使之总体积为500 μL,混匀后37 ℃孵育20 min后加入每孔细胞中,5 h后吸出培养液弃掉,每孔加入2 mL DMEM高糖培养基和100 mL/L胎牛血清的培养液。转染后数小时,将6孔板直接放在倒置荧光显微镜下观察,能观察到绿色荧光与目的基因的融合蛋白开始表达,转染后24 h后,用2.5 g/L胰酶消化细胞,PBS重新悬浮细胞,调整细胞密度大约为5×105/mL左右,流式细胞仪检测转染效率。
1.7 目的基因表达的鉴定 Trizol法提取细胞中的总RNA。在微量核酸/蛋白定量仪上定量,10 g/L琼脂糖凝胶鉴定质量。按照RevertAidTMFirst Strand cDNA Synthesis Kit试剂盒将提取的总RNA中的mRNA反转录为cDNA并以此为模板,用上述PCR体系对所转染的硒蛋白基因进行扩增,并10 g/L琼脂糖凝胶电泳鉴定。
2 结 果
2.1 SelS基因克隆的鉴定结果 构建了SelS和trun-SelS基因的克隆,琼脂糖电泳凝胶图显示(图2),重组质粒经双酶切后,均释放出了与目的硒蛋白DNA片段大小一致的DNA片段和载体pEGFP-C1,与测序结果的BLAST比对并确定插入片段碱基无缺失、无插入、无突变,一致率为100%,说明目的克隆已被成功构建。
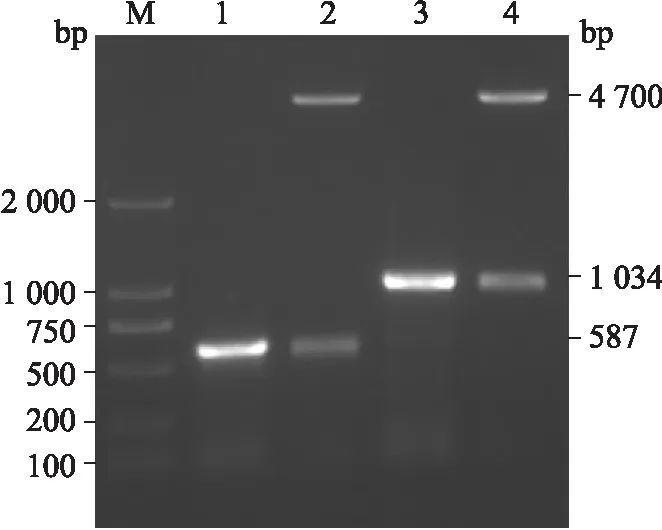
图2 SelS和trun-SelS基因重组质粒的琼脂糖凝胶电泳图
Fig.2 Agarose gel electrophoresis of recombinant plasmids of selenoprotein S genes
M:DNA marker(D2000);1:trun-SelS的PCR产物;2:trun-SelS重组质粒的双酶切产物;3:为SelS的PCR产物;4:SelS重组质粒的双酶切产物。
2.2 转染后绿色荧光蛋白的表达 转染trun-SelS和SelS 24 h后,绿色荧光蛋白表达很好(图3),将细胞消化后上流式细胞仪检测,转染效率达到40%以上。这说明目的蛋白成功表达。
2.3 转染24 h时RT-PCR检测目的硒蛋白基因的表达情况 trun-SelS组和SelS组的PCR产物的电泳结果都出现了明显的trun-SelS基因条带,而mock组和空载体组由于目的基因拷贝数低,没有出现明显的目的条带,说明转染成功,即被转染的细胞高表达了trun-SelS基因;由于SelS的基因片段中包含trun-SelS基因片段,所以在SelS组中也有trun-SelS基因片段的高表达;由于trun-SelS的基因片段中没有完全包含SelS基因片段,所以PCR产物的电泳结果只有SelS组高表达了SelS目的基因,也说明了SelS组重组质粒的成功转染;4组PCR产物均出现了明显的内参基因条带,且灰度基本一致,说明4组12个样品的均一性很好,比较是有意义的,结果真实可靠(图4)。
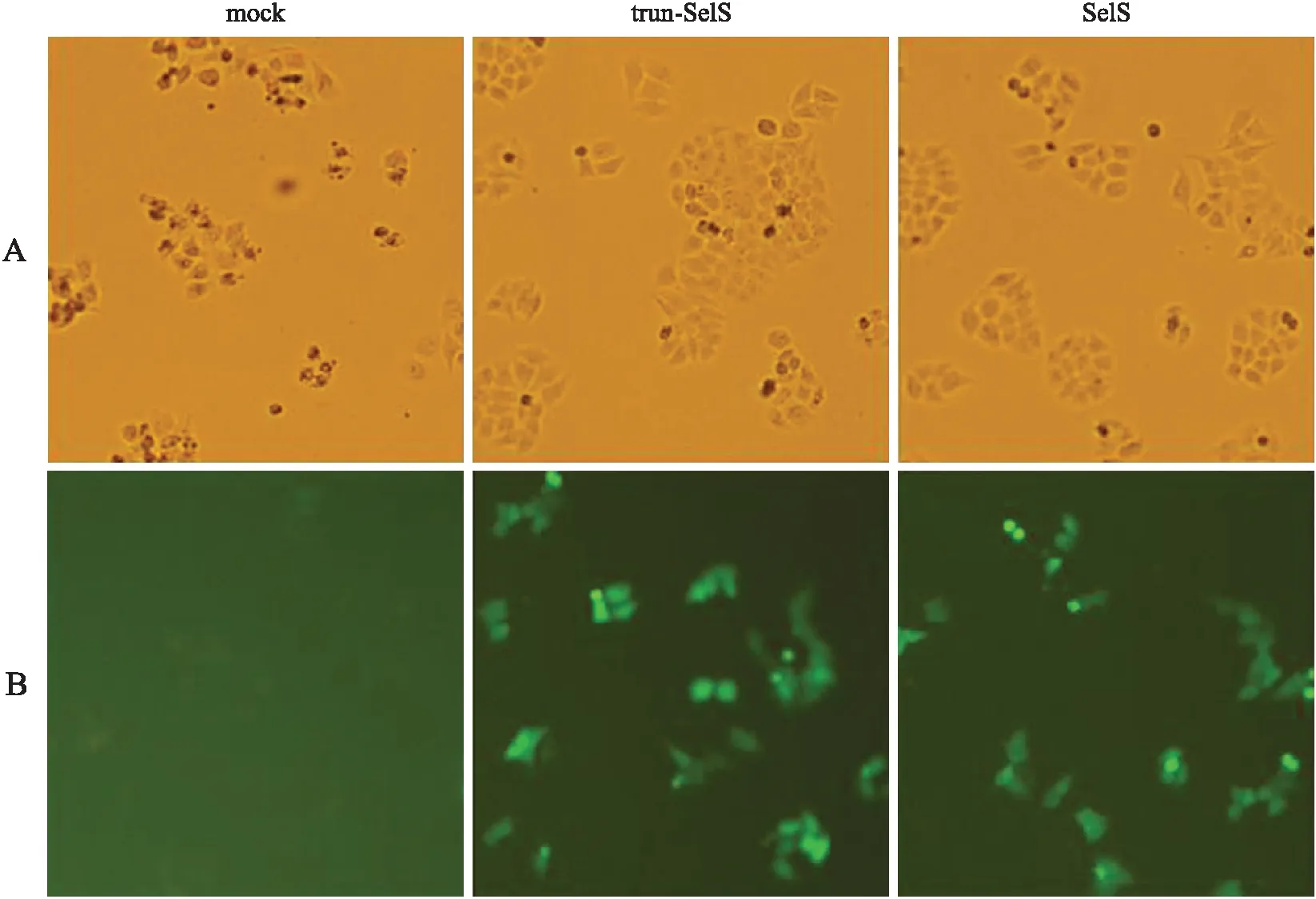
图3 硒蛋白S截短和全长基因转染进Hela细胞24 h绿色荧光蛋白的表达
Fig.3 GFP expression of normal and truncated selenoprotein S genes at 24 hours after transfection into Hela cells
A:细胞在光镜下形态;B:细胞在荧光显微镜下形态及绿色荧光蛋白的表达。
这说明已经成功转染了SelS截短和正常的基因构建体,并使目的基因在细胞系中得以高表达,可进一步观察这些目的基因高表达后对细胞的生物学作用。
3 讨 论
绿色荧光蛋白由于其易表达、分子质量小、蛋白稳定及对细胞无毒性等优点, 是近年来在分子生物学中应用最广泛的标记性蛋白质之一。绿色荧光真核表达载体pEGFP-C1作为改良后的一种变体,更易于观测和研究目的基因的表达、调控及其目的蛋白在生物体内定位和信号转导等。
应用此载体,我们成功构建了SelS截短和正常的基因重组质粒,截短体基因只包含mRNA的CDS,全长包括mRNA的CDS和3′UTR序列。在硒蛋白mRNA的3′TUR中存在SECIS,该结构在翻译过程中能将UGA翻译为Sec,硒蛋白中的硒都以Sec位于活性中心发挥其生物学作用[2]。所以,存在3′TUR的硒蛋白基因将表达出正常的有酶活性的SelS,而仅含CDS序列的硒蛋白基因将表达出羧基端缺失-Sec-Gly的无酶活性的截短SelS。

图4 转染硒蛋白S截短和全长基因后RT-PCR产物的的电泳结果
Fig.4 RT-PCR results of normal and truncated selenoprotein S genes after plasmid transfection
A图中1~3:mock组;4~6:空载体组;7~9:trun-SelS组;10~12:SelS组;每组中设3个平行样;B:内参GAPDH。
ABESTAL[8]做了有关截短硒蛋白的研究:利用化学技术去除掉蛋白质肽链上最后两个氨基酸-Sec-Gly,制作出截短的硒蛋白,然后测定其酶活性,发现截短硫氧还蛋白还原酶1(TrxR1)的自身酶活性与正常全长相比非常低,几乎检测不到,硒含量也接近无。研究者利用BioPORTER技术将这种截短的硒蛋白导入A549细胞后,发现其与具有正常酶活性TrxR1蛋白比较能引起细胞快速死亡,但机制不清[8]。此研究结果引起我们极大的兴趣。日本学者又研究了TrxR2的剪接体,在其cDNA水平上编码区有3个碱基的缺失、终止密码子与3′UTR的SECIS之间有1228个碱基的插入。为了检测其在细胞内的功能,他们将此种截短的TrxR2剪接体转染至Hela细胞系中并调节其转录,表达出的截短的TrxR2能引起细胞凋亡,凋亡原因为NADPH的还原和细胞内ROS水平的升高。此研究说明截短的硒蛋白在细胞生理病理中起着特殊的调节作用[9]。
鉴于SelS在机体内丰富的表达并有其重要的生物学功能,迄今为止还没有研究通过基因重组将SelS的不同剪接体形式在细胞内表达,而本研究通过将这两种SelS的重组质粒在靶细胞中表达,以便模拟正常和硒缺乏状态下硒蛋白在机体内的生物学效应,为进一步研究低硒或缺硒疾病的致病机制提供实验基础。
[1] LU J, HOLMGREN A. Selenoproteins[J]. J Biol Chem, 2009, 284:723-727.
[2] PAPP LV, LU J, HOLMGREN A, et al. From selenium to selenoproteins: Synthesis, identity, and their role in human health[J]. Antioxid Redox Signal, 2007, 9:775-806.
[3] MISTRY HD, BROUGHTON PIPKIN F, REGMAN CW, et al. Selenium in reproductive health[J]. Am J Obstet Gynecol, 2012, 206:21-30.
[4] LIN HC, HO SC, CHEN YY, et al. CRL2 aids elimination of truncated selenoproteins produced by failed UGA/Sec decoding[J]. Science, 2015, 349(6243):91-95.
[5] HOWARD MT, CARLSON BA, ANDERSON CB, et al. Translational redefinition of UGA codons is regulated by selenium availability[J]. J Biol Chem, 2013, 288:19401-19413.
[6] MINIARD AC, MIDDLETON LM, BUDIMAN ME, et al. Nucleolin binds to a subset of selenoprotein mRNAs and regulates their expression[J]. Nucleic Acids Res, 2010, 38:4807-4820.
[7] BUBENIK JL, MINIARD AC, DRISCOLL DM. Alternative transcripts and 3’UTR elements govern the incorporation of selenocysteine into selenoprotein S[J]. PLoS One, 2013, 8:e62102.
[8] ABESTAL K, ARNER ES. Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine[J]. J Biol Chem, 2003, 278:15966-15972.
[9] CHANG EY, SON SK, KO HS, et al. Induction of apoptosis by the overexpression of an alternative splicing variant of mitochondrial thioredoxin reductase[J]. Free Radic Biol Med, 2005, 39:1666-1675.
