SLC37A4基因新突变致糖原累积病Ⅰb型一家系报道并文献复习
2018-02-15张远达董青伟张少辉张瑜桑艳梅丁召路
张远达,董青伟,张少辉,张瑜,桑艳梅,丁召路
糖原累积病(glycogen storage disease,GSD)是一种以糖原代谢异常为主要特征的常染色体隐性遗传病,主要临床表现为肝脏增大、高乳酸血症、高尿酸血症、高脂血症、低糖血症、生长发育迟缓、中性粒细胞计数减少、中性粒细胞功能损伤及炎症性肠病等[1];根据转运体或酶缺陷类型可将GSD分为12种亚型,其中糖原累积病Ⅰ型(glycogen storage disease type Ⅰ,GSDⅠ)最为常见,主要由葡萄糖-6-磷酸酶α(G6P-α)/葡萄糖-6-磷酸酶转移酶(G6PT)复合物活性缺陷引起。GSDⅠ主要分为GSDⅠa型和GSDⅠb型两种类型,其中GSDⅠa型约占80%,GSDⅠb型约占20%[2]。研究表明,GSDⅠb型主要由编码G6PT的SLC37A4基因突变所致[3],目前我国已报道32例SLC37A4基因突变患者,但大部分患者仅进行了基因测序,并未进行肝脏穿刺病理学检查。本文报道了GSDⅠb型一家系,分析了其临床特征、肝脏穿刺病理学检查结果及SLC37A4基因测序结果,并进行了文献复习,旨在提高临床医师对GSDⅠb型的认识。
1 病例简介
1.1 先证者 女,6岁5个月,因“间断鼻出血5年”于2017年5月入住保定市儿童医院血液科。患儿于5年前无明显诱因出现间断鼻出血,发作频率为1次/1~3个月。患儿为足月顺产,第1胎,智力发育正常,患儿父母非近亲结婚,母亲第2胎为双胞胎男孩。查体:体质量为19 kg(<参考范围P3),身高为106.0 cm(<参考范围P3),娃娃脸面容,轻度贫血貌,皮肤无皮疹及出血点,心肺及神经系统查体未见异常,腹部膨隆,肝脏肋下8 cm、剑突下10 cm可触及且质地中等,脾脏肋下未触及。
实验室检查:白细胞计数为4×109/L〔参考范围(4~10)×109/L〕,红细胞计数为3.7×1012/L〔参考范围(3.5~5.5)×1012/L〕,血小板计数为275×109/L〔参考范围(100~400)×109/L〕,血红蛋白为96 g/L(参考范围110~160 g/L),中性粒细胞计数为1.3×109/L〔参考范围(1.4~6.5)×109/L〕,丙氨酸氨基转移酶为32 U/L(参考范围5~40 U/L),天冬氨酸氨基转移酶为42 U/L(参考范围5~40 U/L),三酰甘油为2.70 mmoL/L(参考范围0.40~1.70 mmol/L),尿酸为654 μmol/L(参考范围119~416 μmol/L),乳酸为11 μmol/L(参考范围0~2 μmol/L),空腹血糖为3.4 mmol/L(参考范围3.9~6.1 mmol/L);嗜肝病毒及肝炎病毒阴性;骨髓穿刺结果未见异常;腹部彩超检查结果示肝右肋下8.0 cm、剑突下9.5 cm、左肋下6.0 cm,脾未探及肿大;腹部CT检查结果示肝脏轮廓增大,密度稍增高,左肾稍大。
1.2 先证者大弟 男,2岁11个月,2年前出现间断鼻出血。查体:体质量为14 kg(介于参考范围P10~P25),身高为95.0 cm(介于参考范围P10~P25),中度贫血貌,娃娃脸面容,腹部膨隆,肝脏肋下8 cm、剑突下9 cm可触及且质地中等,脾脏肋下3 cm可触及;余查体未见明显异常。
实验室检查:白细胞计数为5×109/L,红细胞计数为2.9×1012/L,血小板计数为339×109/L,血红蛋白为71 g/L,中性粒细胞计数为1.05×109/L〔参考范围(0.72~4.60)×109/L〕,丙氨酸氨基转移酶为101 U/L,天冬氨酸氨基转氨酶为105 U/L,三酰甘油为2.59 mmol/L,尿酸为657 μmol/L,乳酸为11 mmol/L,空腹血糖为4.3 mmol/L;骨髓穿刺结果未见异常;腹部彩超检查结果示肝右肋下8.0 cm、剑突下9.0 cm并可见一枚内部小强回声结节(提示肝腺瘤),脾左肋下2.7 cm。
1.3 先证者二弟 男,2岁11个月,2年前出现间断鼻出血并于1年前因贫血而进行输血治疗,输血量不详。查体:体质量为14 kg(介于参考范围P10~P25),身高为94.5 cm(介于参考范围P10~P25),中度贫血貌,娃娃脸面容,腹部膨隆,肝脏肋下12 cm、剑突下7 cm可触及且质地中等,脾脏肋下未触及;余查体未见明显异常。
实验室检查:白细胞计数为5×109/L,红细胞计数为3.6×1012/L,血小板计数为212×109/L,血红蛋白为89 g/L,中性粒细胞计数为0.97×109/L〔参考范围(0.72~4.60)×109/L〕,丙氨酸氨基转氨酶为73 U/L,天冬氨酸氨基转氨酶为104 U/L,三酰甘油为2.54 mmol/L,尿酸为636 μmol/L,乳酸为10 mmol/L,空腹血糖为4.2 mmol/L;骨髓穿刺结果提示三系增生骨髓象;腹部彩超检查结果示肝右肋下12.6 cm、剑突下6.7 cm、左肋下3.6 cm且回声不均匀、可见多个低回声结节(最大者为0.9 cm×0.6 cm,提示肝腺瘤),脾左肋下1.7 cm且实质回声均匀。
1.4 先证者母亲 女,28岁,既往体健,孕2产3,其中孕1产1为先证者,孕2产2为先证者大弟,孕2产3为先证者二弟;查体未见异常,血常规、肝功能、空腹血糖、血脂指标、尿酸、乳酸等实验室检查指标均正常;腹部彩超检查结果示肝脾大小、结构均正常。
1.5 先证者父亲 男,30岁,既往体健;查体、实验室检查结果及腹部B超均未见异常。
1.6 家系基因分析 征得患儿父母知情同意后采集先证者、先证者两个双胞胎弟弟及先证者父母血标本并送北京迈基诺医学检验所进行基因检测,结果示先证者、先证者两个双胞胎弟弟及先证者母亲SLC37A4基因c.273C>A(p.N91K)突变(杂合突变),即c.273位点胞嘧啶被腺嘌呤所取代,而由其编码的第91个氨基酸由天冬酰胺变为赖氨酸,为新发突变,而先证者父亲基因型未发现异常(见图1~2);经二代测序法及多重连接探针扩增技术(MLPA)验证,未发现内含子突变、大片段缺失或插入,与既往文献报道的基因突变类型不同。
1.7 肝脏穿刺病理学检查结果由于先证者母亲检出相同突变基因而先证者父亲未检出,因此依据基因测序结果无法确诊,征得先证者父母知情同意后进一步对先证者进行肝脏穿刺病理学检查,其中光镜检查结果示肝细胞肿胀、细胞核增大并可见糖原核,肝细胞镶嵌排列,汇管区纤维组织增生,散在淋巴细胞,少量嗜酸粒细胞浸润,PAS 染色阳性(见图3);电镜检查结果示肝板排列略有不整,肝细胞高度肿胀,核圆形且其内无糖原颗粒沉积,胞质内电子密度低,可见片状空区,其间散在糖原颗粒,线粒体大小较一致,多分布于核周及胞膜下,脂肪变性明显,可见较多大小不一的脂滴,毛细胆管结构完整,未见纤维化改变(见图4)。结合光镜检查结果及PAS染色结果,先证者肝脏穿刺病理学检查结果符合GSD超微结构改变。
1.8 诊断及治疗 根据患儿临床症状、实验室检查结果及肝脏穿刺病理学检查结果确诊为GSD,结合基因测序结果考虑为GSDⅠb型;给予生玉米淀粉口服并嘱患儿父母保证患儿碳水化合物、脂肪、蛋白质的均衡摄入。截至发稿,患儿病情平稳,继续随访中。
2 文献复习
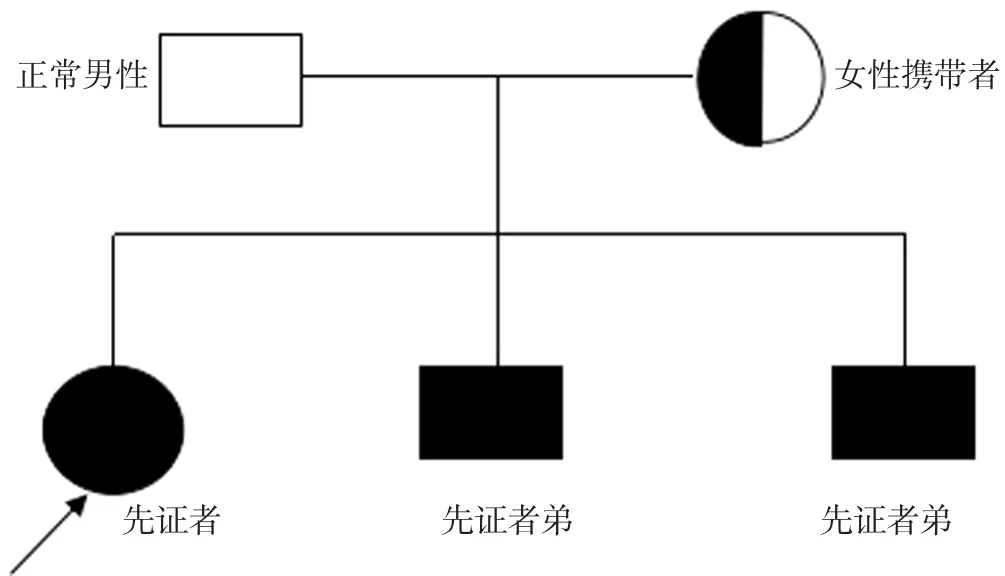
图1 GSDⅠb型患者家系图Figure 1 Family tree of GSDⅠtype b
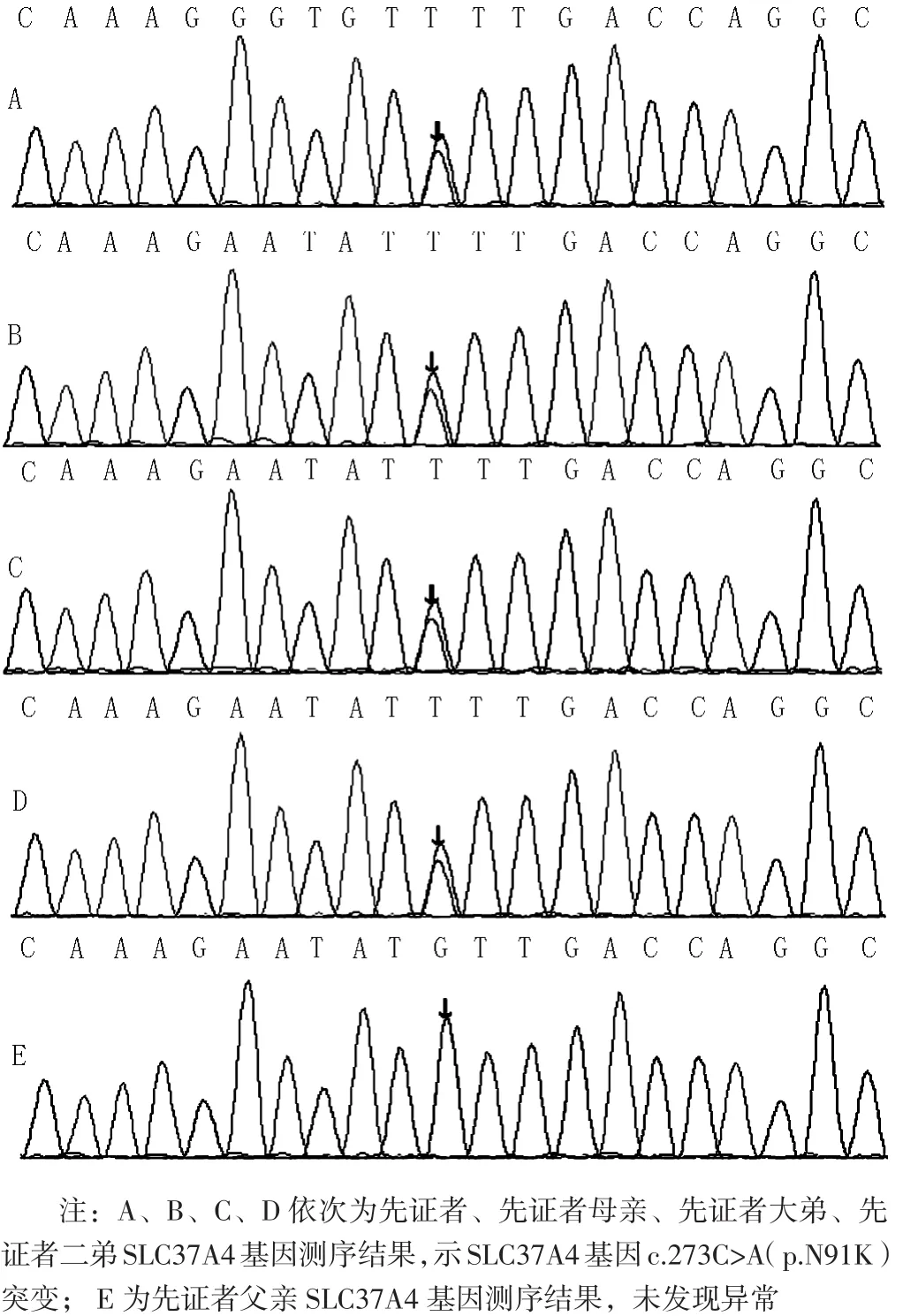
图2 GSDⅠb型一家系SLC37A4基因测序结果Figure 2 Gene sequencing results of the family with GSDⅠtype b

图3 先证者肝脏穿刺光镜检查结果(PAS 染色,×100)Figure 3 Light microscopy results of liver biopsy of the proband
以“糖原累积病Ⅰb型”“glycogen storage disease typeⅠb”“SLC37A4”为检索词检索中国知网(CNKI)、万方数据知识服务平台、PubMed等数据库,检索时间为建库至2017年10月,共检索到138例GSDⅠb型病例,其中临床资料较完整者105例。
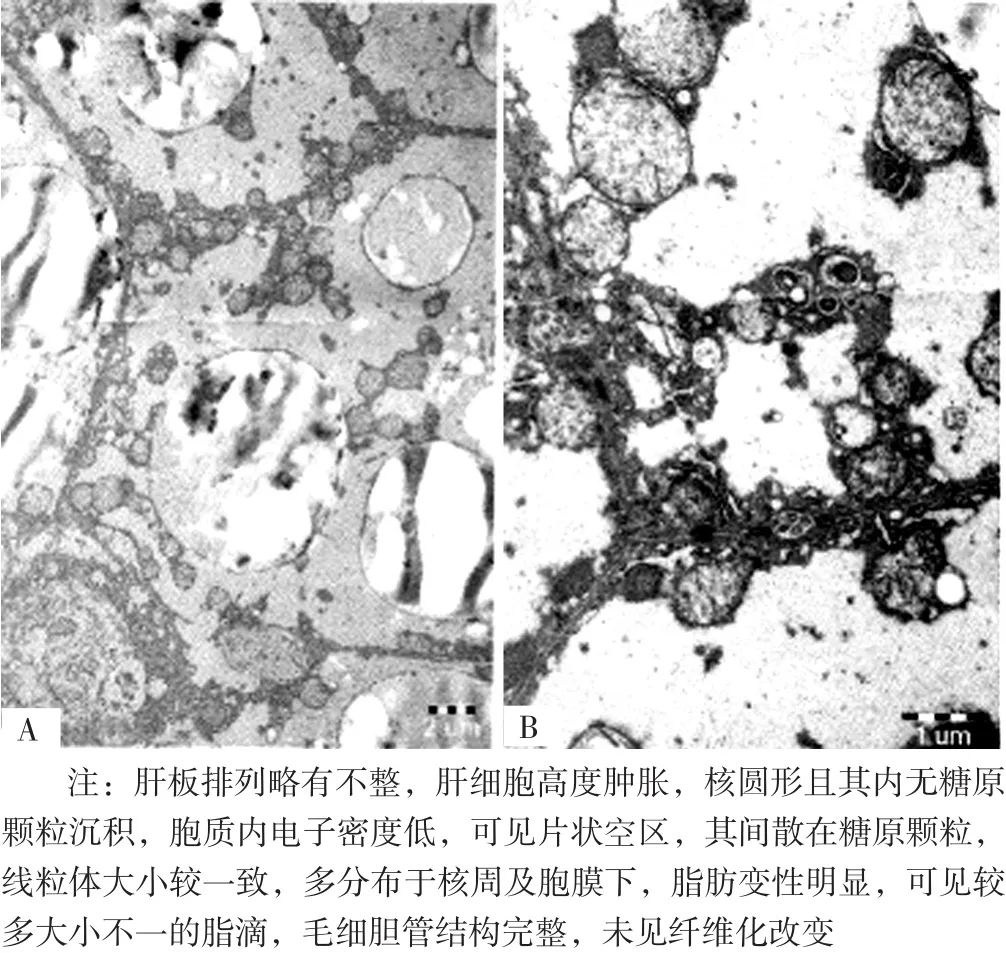
图4 先证者肝脏穿刺电镜检查结果(×10 000)Figure 4 Result of liver puncture electron microscopy of proband
2.1 临床表现 105例临床资料较完整者中肝大105例(占100.0%),中性粒细胞计数降低101例(占96.2%),低糖血症88例(占83.8%),高乳酸血症64例(占60.9%),高三酰甘油血症39例(占37.1%),高尿酸血症33例(占31.4%),炎症性肠病9例(占8.6%)。
2.2 基因突变类型 结合人类基因突变数据库(HGMD),共检索到110种SLC37A4基因突变类型,其中错义突变60种,小片段缺失19种,剪切突变17种,无义突变12种,小片段插入8种,大片段缺失2种,小片段插入缺失2种。
2.3 来自中国的病例 中国地区报道了32例SLC37A4基因突变病例,其中临床资料较完整者31例。31例临床资料较完整的SLC37A4基因突变病例中男19例,女12例;30例采用基因诊断,1例经肝脏穿刺病理学检查明确诊断;临床表现:肝大31例(占100.0%),低糖血症30例(占96.8%),中性粒细胞计数降低30例(占96.8%),高乳酸血症30例(占96.8%),高尿酸血症30例(占96.8%),高三酰甘油血症30例(占96.8%),肝功能异常29例(占93.5%),反复感染18例(占58.1%),口腔溃疡9例(占29.0%),关节症状5例(占16.1%),炎症性肠病2例(占6.5%);涉及16种基因突变类型:错义突变(包括p.Pro191Leu、p.Leu23Arg、p.Gly281Val、p.Gly115Arg、p.Tyr24His、 p.Gly149Glu、p.Trp246Arg、p.Arg415Gly)8种,缺失突变(包括c.1042_1043 del CT、c.1014_1120del107、IVS8+1_4delGTAA、c.354_355insC)4种,剪切突变(c.870+5G>A、c.784+1G>A)2种,移码突变(c.959~960 insT)1 种,无义突变(p.Arg415X)1 种[4~12]。
3 讨论
目前,临床主要通过基因检测和/或肝脏穿刺病理学检查诊断GSDⅠ,其中基因检测因具有无创性而成为诊断GSDⅠ的首选[3],而由于GSDⅠa型和GSDⅠb型患者临床表现相似,因此二者的鉴别诊断亦主要依据基因检测。GSDⅠa型主要由G6PC基因突变导致葡萄糖-6-磷酸酶(G6P)缺乏而引发,典型临床表现为肝肾肿大、低糖血症、生长发育迟缓、血小板功能障碍、高乳酸血症、高尿酸血症、高脂血症、肝脏腺瘤、痛风、骨质疏松及肾功能不全等[13]。GSDⅠb型主要由编码G6PT的SLC37A4基因突变导致G6PT缺乏而引发[14],患者除具有GSDⅠa型临床表现外,还会出现中性粒细胞、单核细胞功能损伤及慢性中性粒细胞计数减少,表现为反复的细菌感染及口腔、肠道黏膜溃疡等[3]。本文中先证者及其双胞胎弟弟均以鼻出血为首发症状,查体可见娃娃脸、腹部膨隆,实验室检查发现乳酸性酸中毒、高尿酸血症、高三酰甘油血症,其中先证者还存在低糖血症、中性粒细胞计数降低、身材矮小等,符合GSDⅠ临床表现。
SLC37A4基因定位于常染色体11q23,包含9个外显子,在肝脏、肾脏、血液、骨骼肌、肠道广泛表达[15]。SLC37A4基因突变可引起其所编码的G6PT缺乏,导致葡萄糖-6-磷酸(G6P)转化为葡萄糖出现障碍,进而引发一系列临床症状,因此临床常对SLC37A4基因进行测序而诊断GSDⅠb型[1]。目前研究认为,GSD是一种常染色体隐性遗传病,有关于GSDⅠa型患者仅携带有1个基因杂合突变位点的报道[3],但尚未见GSDⅠb型患者仅携带有1个基因杂合突变位点(GSDⅠb型常染色体显性遗传)的报道。王艳等[13]曾报道1例GSDⅠa型患儿及其母亲G6PC基因同位点杂合突变,但患儿父亲及姐姐未检出该位点突变,因此考虑为杂合突变所致GSDⅠa表型的特例。本文中先证者、先证者两个双胞胎弟弟及先证者母亲经基因测序证实SLC37A4基因杂合突变,但先证者父亲未检出该位点突变,且本家系未发现内含子突变、大片段缺失或插入,这与目前已知的GSDⅠ常染色体隐性遗传方式不同,但尚不能简单地认为该突变位点单纯由先证者母亲遗传而来,可能与先证者父亲突变位于启动子区域或引物上等而未被发现有关。此外,笔者采用“SLC37A4”在HGMD进行检索后共检索到110种基因突变类型,但c.273C>A(p.N91K)未检出,证实为新发突变。
值得强调的是,基因检测不能明确诊断GSD时应积极行肝脏穿刺病理学检查以明确诊断[1]。本家系经基因检测不能明确诊断,遂对先证者进行肝脏穿刺病理学检查,最终确诊为GSD,结合基因检测结果考虑本家系先证者及其两个弟弟均为GSDⅠb型。需要指出的是,目前研究认为GSDⅠb型患者基因型与表型之间并无明显相关性,本文中先证者及其两个双胞胎弟弟具有相同基因型及典型GSDⅠ临床表现,而具有相同基因型的先证者母亲却无相应临床表现,同时先证者存在中性粒细胞计数降低但肝脏彩超检查结果无异常,而先证者两个双胞胎弟弟中性粒细胞计数无明显下降但肝脏彩超检查结果均提示肝腺瘤,证实GSDⅠb型患者基因型与表型之间无明显相关性。
综上所述,GSDⅠb型较罕见,本文报道的GSDⅠb型一家系由SLC37A4基因c.273C>A(p.N91K)突变(杂合突变)导致,为新发突变;临床发现具有典型GSD临床表现而疑诊GSD者应首选基因检测,基因检测不能明确诊断者应积极行肝脏穿刺病理学检查以明确诊断。
