热水提取与超声提取的麦冬多糖结构与构象特征的比较研究
2018-01-25王小梅薛红杰郝大鹏孙润广
王小梅,王 妙,薛红杰,张 明,郝大鹏,孙润广
﹙1.西安航空学院 理学院,陕西 西安 710077;2.陕西师范大学 物理学与信息技术学院生物物理与生物医学工程实验室,陕西 西安 710062﹚
麦冬为百合科多年生草本植物,麦冬多糖作为麦冬的主要活性成分之一,具有降血糖、抗缺氧、抗肿瘤、抗辐射及免疫调节作用等多种生物活性[1-4]。为了得到高质量的麦冬多糖,提取方法尤为重要。与传统提取方法相比,超声提取法具有独特的物理化学效应(如机械效应、热效应、空化效应等),可以加速溶剂向植物细胞中渗透,提高提取率,便于提取,已被国内外学者广泛地应用于中药多糖的提取[5-7]。超声提取可以明显提高中药多糖的提取率,但超声提取对多糖结构及生物活性的影响还有待进一步研究。而相对于一级结构,多糖的高级结构[8-9]尤其是溶液性质和链构象对多糖生物活性的作用更大。Tao等[10]研究发现,具有三螺旋结构的裂褶菌多糖有较高的抗肿瘤活性,而具有相似一级结构的单螺旋β-葡聚糖却无此活性。且研究发现[11-12],几十种具有降血糖活性的中药多糖,其一级结构也不一定相同。这充分说明多糖高级结构对其生物活性的影响更为显著,而多糖的溶液行为与链构象直接影响其活性的表达。因此,为了将超声更好地应用到中药多糖提取中,研究超声对中药多糖高级结构尤其是溶液行为和链构象的影响至关重要。本文利用热水与超声提取麦冬多糖,并对其结构和构象特征进行比较研究,揭示了超声对麦冬多糖结构及溶液构象的影响,为将超声更好地应用到中药有效成分的提取中提供理论和实验依据。
1 实验部分
1.1 材料、试剂与仪器
麦冬,产地湖北,购于西安市万寿路中药材市场。95%乙醇、苯酚、氯化钠、氢氧化钠、浓硫酸均为分析纯,DEAE-cellulose52纤维素(Whatman公司),Sephacryl S-300葡聚糖凝胶(Pharmacia公司),系列层析柱(2.5×60 cm,上海琪特分析仪器有限公司)。
FD-1A真空冷冻干燥机、磁力搅拌器、TU-1810紫外-可见分光光度计(北京普析通用仪器有限责任公司);RE-52AA型旋转蒸发器、Chirascan型圆二色谱仪(英国应用光物理公司);SPM-9500J3型原子力显微镜(日本岛津公司)。
1.2 麦冬多糖的提取及纯化
将麦冬块根60 ℃烘干,粉碎,加入4倍体积的95%乙醇脱脂(3次),烘干,分别利用热水与超声两种方法提取麦冬多糖。热水提取:80 g脱脂后的粉末,以料液比1∶10加蒸馏水,80 ℃恒温水浴搅拌提取2 h(重复提取2次);超声提取:80 g脱脂后的粉末,按料液比1∶10加蒸馏水并搅拌使其充分溶解,利用超声波细胞粉碎机提取。提取条件:参考文献[13],选取超声功率400 W,超声时间15 s,间隔时间15 s,超声次数90次(重复提取2次)。将两种方法提取得到的提取液减压浓缩,用4倍体积的95%乙醇醇沉,离心、透析、真空干燥后得水提麦冬粗多糖WPOJ与超声提取麦冬粗多糖UPOJ。紫外-可见分光光度计检测,发现WPOJ与UPOJ在260~280 nm均无特征吸收峰,说明WPOJ及UPOJ均不含蛋白质和核酸。
对WPOJ与UPOJ分别进行DEAE-cellulose52纤维素柱层析,洗脱液分别为蒸馏水、0.01 mol/L NaCl、0.03 mol/L NaCl,流速6 s/滴,15 min/管,全自动部分收集器收集洗脱液,苯酚-硫酸法在490 nm波长处检测糖含量。以试管数目为横坐标,吸光度值为纵坐标绘制DEAE-cellulose52色谱柱洗脱曲线图。将收集最多的组分进行凝胶色谱分离。流速为5 s/滴,15 min/管,用全自动部分收集器收集洗脱液,苯酚-硫酸法检测。以试管数目为横坐标,吸光度值为纵坐标绘制Sephacryl S-300色谱柱洗脱曲线图。合并各主峰溶液,浓缩,蒸馏水透析,真空冷冻干燥,备用。
1.3 实验方法
1.3.1麦冬多糖WPOJ-DS与UPOJ-DS分子量的测定采用高效液相色谱法对WPOJ-DS与UPOJ-DS的分子量进行测定。色谱柱为Shodex Ohpak SB-804 HQ,检测器为Waters 2410示差折光检测器,流动相为高纯水,流速0.8 mL/min,进样体积20 μL,进样浓度1 mg/mL。以不同标准葡聚糖分子量(200 000、70 000 、40 000、10 000、5 000 Da)的对数为纵坐标,以洗脱峰的保留时间为横坐标,得到线性回归方程。将多糖样品溶于高纯水制成1 mg/mL的溶液,在相同色谱条件下进样分析,记录保留时间,带入回归方程计算麦冬多糖的平均分子量[14]。
1.3.2单糖组成检测将5 mg多糖样品溶于3 mL 2 mol/L三氟乙酸中,110 ℃下完全酸水解。水解产物糖腈乙酰化后进行GC分析。气相色谱条件:色谱柱为OV-17毛细管柱;FID检测器(260 ℃);升温程序:150 ℃(7 ℃/min)→190 ℃(15 ℃/min)→250 ℃;进样口温度280 ℃;进样量1 μL;载气流速10 mL/min,Air∶H2∶N2=300∶30∶30。
1.3.3傅立叶变换红外光谱检测多糖样品与干燥的KBr按照质量比1∶100混合,充分研磨后压片,利用傅立叶变换红外光谱仪在4 000~400 cm-1范围内进行扫描。
1.3.4部分酸水解25 mg多糖样品中加入0.02 mol/L的TFA 5 mL,110 ℃下水解3 h,用蒸馏法除去多余的TFA后以蒸馏水透析48 h,得到透析袋内外不同分子量截留液,分别冷冻干燥,完全酸水解,糖腈乙酰化后进行GC分析。
1.3.5甲基化分析采用改良的Hakomori法[15]进行甲基化,用红外光谱在3 600~3 300 cm-1检测有无吸收峰,以确定甲基化是否完全。将完全甲基化的多糖进行酸水解,水解产物乙酰化后进行GC-MS分析。分析条件:SE-54石英毛细管柱(15 m×0.25 mm×0.25 μm);程序升温:100 ℃(保温6 min)→280 ℃(保温8 min),升温速率20 ℃/min,进样口温度280 ℃,载气:He,流速:12 mL/min,离子源:EI,70 eV。
1.3.6核磁共振分析5 mg多糖样品溶于0.5 mL重水(D2O)中,制成10 mg/mL的溶液,用AVANCE-Ⅲ 400M 核磁共振仪对其进行1H NMR 分析,测定温度40 ℃(313 K)。取干燥多糖样品溶于重水(D2O) 中,使其达到饱和状态,用AVANCE-Ⅲ 400 M 核磁共振仪对其进行13C NMR分析,测定温度40 ℃(313 K)。
1.3.7刚果红实验配制样品多糖(WPOJ-DS与UPOJ-DS)溶液5 mg/mL、NaOH溶液1 mol/L、刚果红溶液80 μmol/L,将2 mL多糖溶液与相同体积刚果红溶液混合,加入NaOH溶液,使NaOH的最终浓度由0~0.5 mol/L等梯度增加,静置10 min,利用紫外-可见分光光度计于400~600 nm波长处进行光谱扫描。以刚果红为对照,测定各碱性条件下的最大吸收波长λ[16]。以NaOH的最终浓度为横坐标,最大吸收波长λ为纵坐标,绘制标准曲线[17]。
1.3.8圆二色谱(CD)实验配制样品多糖溶液3.2 mg/mL,采用Chirascan型圆二色谱仪在180~320 nm波长范围内检测多糖的圆二色性。通过改变外界因素(如添加络合剂、金属离子、溶剂二甲基亚砜及样品多糖溶液的pH值),观察麦冬多糖WPOJ-DS与UPOJ-DS的圆二色谱变化,进而对其构象的变化进行比较分析[18-19]。
1.3.9原子力显微镜观测分别精确称取0.5 mg麦冬多糖WPOJ-DS与UPOJ-DS,用蒸馏水配成25 μg/mL的多糖溶液,磁力搅拌4 h,使其充分溶解。均用蒸馏水稀释至10 μg/mL,分别吸取5 μL滴在新剥离的云母表面上,风干。室温下采用SPM 9500J3型原子力显微镜对麦冬多糖的表面形貌进行扫描观测。图像在contact模式下获得,探针为Si3N4(微悬臂长200 μm,弹性系数0.12 N/m)。AFM图像的形态学特征(如高度、宽度等)采用AFM附带的软件进行分析。
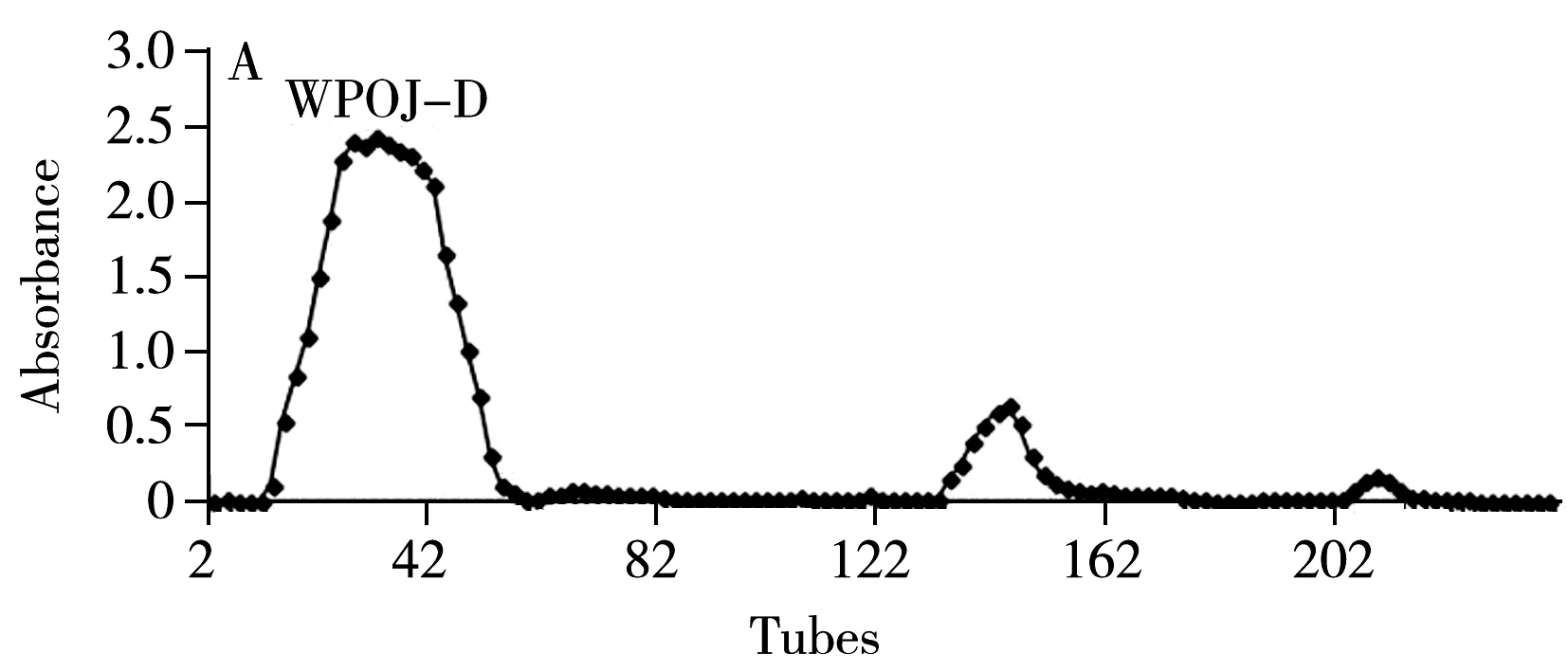

图1 WPOJ(A)与UPOJ(B)经DEAE-52纤维素柱层析洗脱图Fig.1 DEAE-52 cellulose column chromatograms of WPOJ(A) and UPOJ(B)
2 结果与讨论
2.1 分离纯化结果
麦冬多糖经过DEAE-cellulose52纤维素柱的层析纯化洗脱图见图1。图1A为WPOJ的洗脱图,其中第一个大峰为水洗组分,命名为WPOJ-D,第二个峰与第三个峰分别为0.01 mol/L NaCl与0.03 mol/L NaCl洗脱组分。图1B为UPOJ的洗脱图,其中第一个小峰和第二个大峰(命名为UPOJ-D)为水洗组分,第三个峰与第四个峰分别为0.01 mol/L NaCl与0.03 mol/L NaCl洗脱组分。WPOJ-D与UPOJ-D经Sephacryl S-300凝胶柱层析,结果均得到单一对称峰,说明经DEAE-cellulose 52纤维素柱和Sephacryl S-300凝胶柱层析得到了单一的多糖组分,分别命名为WPOJ-DS与UPOJ-DS。
2.2 分子量与单糖组成的检测结果
标准葡聚糖的线性回归方程为lgMw=-0.626x+9.673(r2=0.999 5),式中,x为保留时间(min);Mw为平均分子量。高效液相色谱洗脱结果显示,WPOJ-DS与UPOJ-DS均为单一对称峰,说明两种方法得到的麦冬多糖纯度均较高,出峰时间分别为9.847 min和9.754 min,根据回归方程计算得WPOJ-DS与UPOJ-DS的分子量分别为3 230 Da和3 690 Da。因此,超声提取的麦冬多糖UPOJ-DS的分子量略大于热水提取的麦冬多糖WPOJ-DS。
气相色谱检测结果表明:WPOJ-DS与UPOJ-DS的单糖组分相同,均为果糖、阿拉伯糖和葡萄糖,但摩尔比不同。WPOJ-DS的果糖、阿拉伯糖、葡萄糖的摩尔比为2.68∶7.38∶10.97,而UPOJ-DS为3.40∶8.43∶12.22。说明超声对麦冬多糖单糖组成的摩尔比有影响。
2.3 红外光谱分析结果
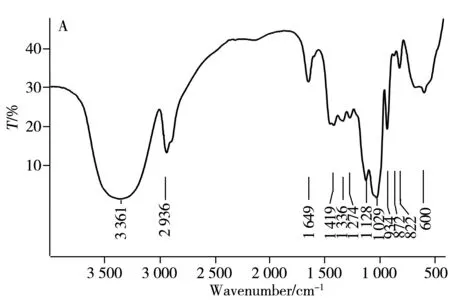
图2为麦冬多糖WPOJ-DS及UPOJ-DS的红外光谱扫描图,从图中可以看出,两种多糖均具有多糖的特征吸收峰,且图2A中934 cm-1处和图2B中935 cm-1处为呋喃环的对称伸缩振动峰;400~600 cm-1处为吡喃环的特征吸收峰[20],说明两种多糖既存在呋喃构型又存在吡喃构型;822 cm-1和820 cm-1处的峰说明两种糖均存在α-构型[21];而两种多糖的区别在于,WPOJ-DS在872 cm-1有β-葡萄吡喃糖的特征吸收峰,而UPOJ-DS中没有,说明经过超声作用后,这一构型受到影响。因此,热水提取的麦冬多糖WPOJ-DS与超声提取的麦冬多糖UPOJ-DS在结构上存在差异。
2.4 部分酸水解结果
WPOJ-DS与UPOJ-DS经0.02 mol/L TFA水解后得到袋内和袋外两种组分。经GC分析,发现袋内组分含有大量葡萄糖,说明WPOJ-DS与UPOJ-DS的主链主要由葡萄糖组成;袋外组分含有果糖、阿拉伯糖和葡萄糖,说明WPOJ-DS与UPOJ-DS的侧链及末端均由果糖、阿拉伯糖和葡萄糖组成。
2.5 甲基化分析结果
将WPOJ-DS与UPOJ-DS经甲基化、水解、还原及乙酰化后得到的甲基化糖醇乙酸酯进行GC-MS分析,根据不同单糖的主要离子碎片和数据库分析,得出WPOJ-DS与UPOJ-DS糖基的连接方式及各种连接键型的比例(表1)。结果表明:WPOJ-DS与UPOJ-DS中1→6葡萄糖为主要键型,可能构成主链,1→3,6葡萄糖为主链的分支点,且由摩尔比可知UPOJ-DS比WPOJ-DS分支点多,且UPOJ-DS中→2)-D-Fruf(1→连接方式的摩尔比较高,说明→2)-D-Fruf(1→很可能位于支链;1→阿拉伯糖位于WPOJ-DS与UPOJ-DS糖链的末端。

表1 WPOJ-DS与UPOJ-DS的甲基化分析结果Table 1 Methylation analysis of WPOJ-DS and UPOJ-DS
(续表1)

Retentiont/minMethylatedsugarLinkagestypesMolarratioofWPOJ-DSMolarratioofUPOJ-DS12.3462,5-Me2-D-Ara→3)-D-Araf(1→2.212.1512.5762,3,4-Me3-D-Glc→6)-D-Glcp(1→8.018.1912.9582,5-Me2-D-Glc→3,6)-D-Glcf(1→3.034.20
2.6 核磁共振分析
麦冬多糖WPOJ-DS与UPOJ-DS的1H NMR图谱表明,两者的异头质子H-1均在δ5.20 ppm左右有1个峰,而一般情况下α型吡喃糖H-1质子的化学位移大于 4.95 ppm,β型吡喃糖H-1质子的化学位移小于4.95 ppm,说明这两种多糖残基主要为α型吡喃糖[22]。麦冬多糖WPOJ-DS与UPOJ-DS的13C NMR如图3所示,13C NMR图中显著的化学位移说明两种多糖都是相对均一的。根据文献[23-24],对麦冬多糖WPOJ-DS的异头碳信号进行归属:δ103.88 ppm为→3)-β-D-Araf(1→异头碳信号;δ103.57 ppm为→6)-β-D-Glcp(1→异头碳信号;δ95.38 ppm为→6)-α-D-Glcp(1→异头碳信号;δ103.71 ppm为→3,6)-α-D-Glcf(1→异头碳信号;δ103.00 ppm为β-D-Araf(1→异头碳信号;δ59.7 ppm为→2)-α-D-Fruf(1→异头碳信号;对麦冬多糖UPOJ-DS的异头碳信号进行归属:δ103.76 ppm为→3)-β-D-Araf(1→异头碳信号;δ97.81 ppm为→6)-α-D-Glcp(1→异头碳信号;δ103.62 ppm为→3,6)-α-D-Glcf(1→异头碳信号;δ103.14 ppm为β-D-Araf(1→异头碳信号;δ59.83 ppm为→2)-α-D-Fruf(1→异头碳信号。因此,WPOJ-DS中存在→6)-β-D-Glcp(1→,而UPOJ-DS中不存在,说明经过超声的作用,该构型受到影响,这与红外光谱观察结果一致。
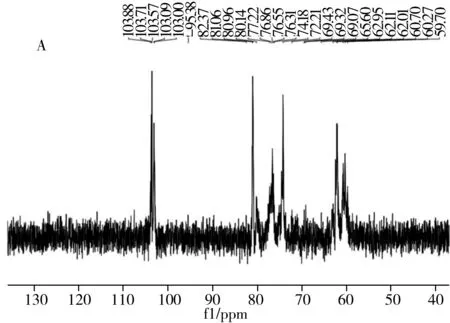
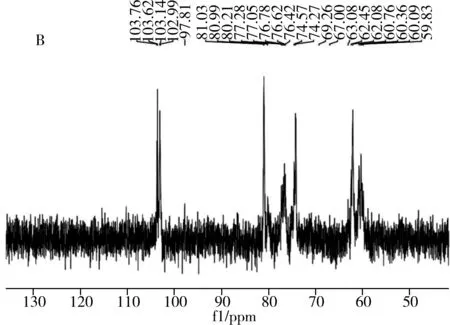
图3 WPOJ-DS(A)与UPOJ-DS(B)的13C NMR图谱Fig.3 The 13C NMR spectra of WPOJ-DS(A) and UPOJ-DS(B)
2.7 刚果红实验结果
考察了麦冬多糖WPOJ-DS与UPOJ-DS分别与刚果红形成的络合物在0~0.5 mol/L NaOH浓度范围内的最大吸收波长变化,结果表明,WPOJ-DS和UPOJ-DS与刚果红络合物的最大吸收波长随NaOH浓度的增大均发生明显红移,当NaOH浓度为0.05~0.2 mol/L时,UPOJ-DS出现亚稳区而WPOJ-DS没有,说明UPOJ-DS存在螺旋结构,对照文献[25]可知,UPOJ-DS分子具有三股螺旋结构。
2.8 圆二色谱测定结果
圆二色谱能判断α螺旋、β折叠及无规线团等信息,常被用于蛋白质和核酸构象的检测。而多糖分子也同样存在不对称结构,尤其在水溶液中其不对称性会更加明显。当外界因素改变时(如金属离子、染料的存在、溶剂种类及酸碱度等变化),必然会引起多糖分子不对称性的变化,因此,可以通过测定多糖的圆二色谱来研究和分析这种变化。
2.8.1比较WPOJ-DS与UPOJ-DS水溶液的CD图谱WPOJ-DS与UPOJ-DS水溶液的CD图谱结果显示,两种多糖的CD图谱均在215 nm左右有负Cotton峰,而UPOJ-DS比WPOJ-DS的负峰略大,说明UPOJ-DS的不对称性略强于WPOJ-DS。这可能与UPOJ-DS和WPOJ-DS的微观结构存在差异有关,且分子量研究结果表明UPOJ-DS的分子量较大,因此,UPOJ-DS分子在水溶液中更容易形成卷曲、折叠和缠绕,表现出分子不对称性增加。
2.8.2络合物对WPOJ-DS与UPOJ-DS溶液构象的影响以CaCl2溶液为溶剂,配制麦冬多糖溶液,进行CD图谱扫描,检测Ca2+对麦冬多糖溶液构象的影响,结果见图4A。从图中可以看出,WPOJ-DS与WPOJ-DS+Ca2+的圆二色谱几乎重合,说明WPOJ-DS不能与Ca2+形成配位结合。而UPOJ-DS+Ca2+相对于WPOJ-DS的圆二色谱负Cotton峰略有增强,说明UPOJ-DS能与Ca2+形成配位结合,使得UPOJ-DS分子间和分子内结合点增多,从而导致分子的不对称性增强。

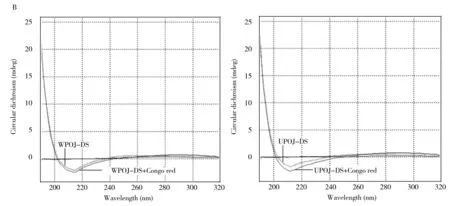
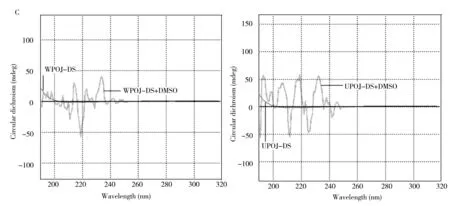
图4 络合物对WPOJ-DS与UPOJ-DS的CD谱的影响Fig.4 Effect of complex on the CD spectrum of WPOJ-DS and UPOJ-DSA:Ca2+;B:Congo red;C:dimethyl sulfoxide
用刚果红溶液作为溶剂,配制麦冬多糖溶液,进行CD图谱扫描,考察染料对麦冬多糖溶液构象的影响,结果见图4B。从图中可以看出,相对于热水提取的麦冬多糖WPOJ-DS,超声提取的麦冬多糖可与刚果红发生明显的复合,使UPOJ-DS分子的不对称性增加,表现为CD谱中负Cotton峰增强。 而刚果红主要与具有螺旋结构的多糖进行络合,此结果说明UPOJ-DS具有螺旋结构,这与刚果红实验结果一致。
以二甲基亚砜(DMSO)作为溶剂配制麦冬多糖溶液,进行CD图谱扫描,考察DMSO对麦冬多糖溶液构象的影响,结果见图4C。从图中可以看出,DMSO作为有机溶剂对WPOJ-DS与UPOJ-DS的构象影响均特别明显,出现较多的正Cotton峰与负Cotton峰,说明DMSO可使WPOJ-DS与UPOJ-DS的不对称性显著增加。
2.8.3不同pH值对WPOJ-DS与UPOJ-DS溶液构象的影响不同pH值(2.0、7.0、12.0)条件下麦冬多糖WPOJ-DS与UPOJ-DS的CD图谱显示,在中性和酸性条件下,WPOJ-DS与UPOJ-DS的构象变化均不大。这可能是由于多糖离子基团的带电荷量及解离程度对多糖分子间与分子内的相互作用影响较大,而WPOJ-DS与UPOJ-DS均为中性多糖,不存在糖醛酸,不带有蛋白质与离子基团,所以影响均较小。碱性条件下,两种多糖的构象变化均较明显,WPOJ-DS与UPOJ-DS在205 nm的吸收峰均发生红移,且UPOJ-DS在190 nm出现新的吸收峰。这说明两种多糖分子间或分子内氢键在碱性条件可以发生变化,且UPOJ-DS的变化更明显。
2.9 原子力显微镜观测结果

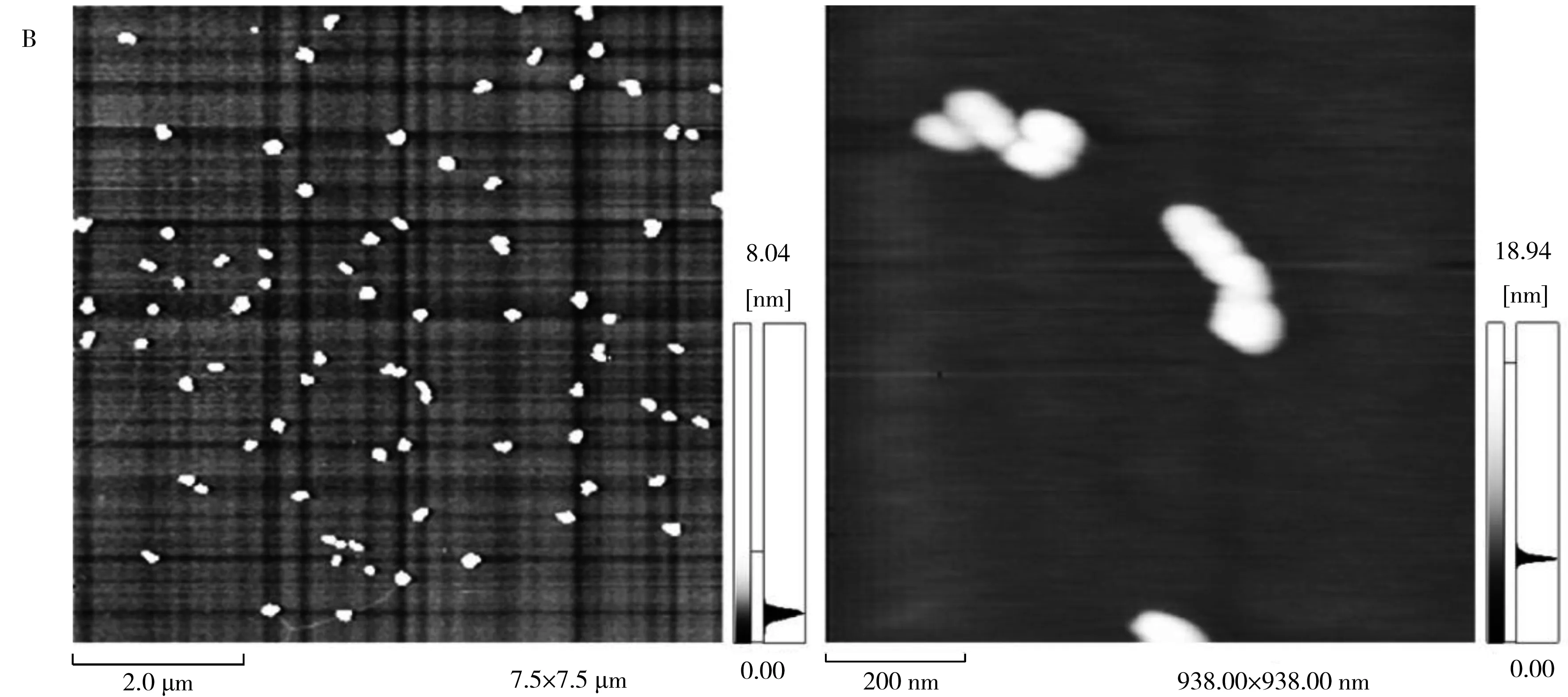
图5 10 μg/mL WPOJ-DS(A)和10 μg/mL UPOJ-DS(B)的AFM图像Fig.5 AFM images of WPOJ-DS(A)and UPOJ-DS(B) on the concentration of 10 μg/mL
图5A为10 μg/mL WPOJ-DS的AFM图像。从图中可以看出,WPOJ-DS呈现出有分支的细丝状,链宽50 nm左右,链高1 nm左右,链长达几个微米,而一般多糖单链的直径约1 nm,说明WPOJ-DS多糖分子链间相互作用及与云母表面的相互作用力使麦冬多糖以单链状态平铺在云母表面。图5B为10 μg/mL UPOJ-DS的AFM图像。从图中可以看出,UPOJ-DS多糖分子较为分散,呈现出小的螺旋棒状结构,高约几个纳米,宽70 nm左右。因此,超声提取的麦冬多糖相对热水提取的麦冬多糖分子的聚集行为发生了明显的变化。经过超声的作用,多糖分子内作用力增强,分子缠绕变短,多糖链与云母表面之间的相互作用力减弱,使多糖呈现出明显的螺旋结构。这与刚果红实验和圆二色谱实验结果均一致。
3 结 论
利用高效液相色谱法、气相色谱法、红外光谱法、部分酸水解、甲基化分析及核磁共振对热水提取与超声提取的麦冬多糖的结构研究表明:超声提取得到的麦冬多糖的分子量大于热水提取的麦冬多糖;两种方法提取得到的麦冬多糖WPOJ-DS与UPOJ-DS的单糖组成摩尔比存在差异;经过超声作用后,→6)-β-D-Glcp(1→这一构型受到影响。刚果红实验、圆二色谱实验及原子力显微镜实验对热水提取与超声提取麦冬多糖的溶液构象研究结果表明:超声提取的麦冬多糖UPOJ-DS存在螺旋结构,而热水提取的麦冬多糖WPOJ-DS没有,且两者在不同溶液环境中的溶液构象也不相同。
综上所述,超声提取对麦冬多糖的结构及溶液构象会产生影响。而与一级结构相比,多糖的高级结构,尤其是溶液性质和链构象对多糖生物活性的作用更大。因此,进一步深入而系统地研究超声对中药多糖高级结构的影响具有意义。
[1] Fan Y P,Ma X,Ma L,Zhang J,Zhang W M,Song X P.Carbohydr.Polym.,2016,135:110-120.
[2] Sun W J,Hu W J,Meng K,Yang L M,Zhang W M,Song X P,Qu X H,Zhang Y Y,Ma L F,Fan Y P.Int.J.Biol.Macromol.,2016,91:918-925.
[3] Xiong S L,Li A L,Huang N,Lu F,Hou D B.Carbohydr.Polym.,2011,86(3):1273-1280.
[4] Chen X M,Tang J,Xie W Y,Wang J J,Jin J,Ren J,Jin L Q,Lu J X.Carbohydr.Polym.,2013,94(1):378-385.
[5] Maran J P,Priya B.Int.J.Biol.Macromol.,2014,70:530-536.
[6] Aguiló-Aguayo I,Walton J,Vias I,Tiwari B K.LWT-FoodScienceandTechnology,2017,77:92-99.
[7] Zheng Q,Ren D Y,Yang N N,Yang X B.Int.J.Biol.Macromol.,2016,91:856-866.
[8] Zhang L F,Ye X Q,Ding T,Sun X Y,Xu Y T,Liu D H.Ultrason.Sonochem.,2013,20(1):222-231.
[9] Feng L P,Cao Y P,Xu D X,Wang S J,Zhang J.Ultrason.Sonochem.,2017,34:609-615.
[10] Tao Y Z,Zhang R Q,Yang W,Liu H T,Yang H J,Zhao Q H.Carbohydr.Polym.,2015,128:179-187.
[11] Li Q,Li W Z,Gao Q Y,Zu Y X.J.FoodSci.,2017,82(10):2487-2494.
[12] Zhao C,Liao Z S,Wu X Q,Liu Y L,Liu X Y,Lin Z X,Huang Y F,Liu B.J.FoodSci.,2014,79(5):1002-1010.
[13] Wang X M,Sun R G,Zhang J,Chen R,Zhang L N,Qiao J J.J.ShaanxiNorm.Univ.:Nat.Sci.Ed.(王小梅,孙润广,张静,陈睿,张力妮,乔进京.陕西师范大学学报:自然科学版),2013,41(3):68-75.
[14] Malinowska E,Krzyczkowski W,apienis G,Herold F.FoodRes.Int.,2010,43(4):988-995.
[15] Needs P W,Selvendran R R.Carbohydr.Res.,1993,245:1-10.
[16] Yi Y,Zhang M W,Liao S T,Zhang R F,Deng Y Y,Wei Z C,Yang B.Carbohydr.Polym.,2012,87(2):1311-1317.
[17] Zhang Y,Gu M,Wang K P,Chen Z X,Dai L Q,Liu J Y,Zeng F.Fitoterapia,2010,81(8):1163-1170.
[18] Kreisman L S C,Friedman J H,Neaga A,Cobb B A.Glycobiology,2007,17(1):46-55.
[19] Jaton J C,Huser H,Blatt Y,Pecht I.Biochemistry,1975,14(24):5308-5311.
[20] Wu X M,Tu P F.J.AsianNat.Prod.Res.,2005,7(6):823-828.
[21] Barker S A,Bourne E J,Stacey M,Whiffen D H.J.Chem.Soc.,1954:171-176.
[22] Hou C M,Chen W N,Chen Y F,Li W.Nat.Prod.Res.Dev.(侯成敏,陈文宁,陈玉放,李伟.天然产物研究与开发),2012,24:556-561.
[23] Agrawal P K.Phytochemistry,1992,31(10):3307-3330.
[24] Bock K,Pedersen C.AdvancesinCarbohydateChemistryandBiochemistry,1983,41:27-66.
[25] Xie C,Gao X D,Wang J,Liu D,Yao W B.J.Chin.Pharm.Univ.(谢晨,高向东,王坚,刘冬,姚文兵.中国药科大学学报),2004,35(6):576-580.
