共沉淀法制备PdFe/Al2O3脱汞吸附剂
2018-01-25霍启煌齐登辉韩丽娜常丽萍鲍卫仁王建成
霍启煌,齐登辉,叶 亮,韩丽娜,常丽萍,鲍卫仁,王建成
1.太原理工大学煤科学与技术省部共建国家重点实验室培育基地,山西 太原 030024;2.太原理工大学材料科学与工程学院,山西 太原 030024
中国是世界上最大的煤炭生产与消费国,我国煤炭主要用于燃烧及传统煤化工行业,而这些行业都存在着资源利用率较低和环境污染严重等问题。因此,大力发展洁净煤技术是解决我国能源与环境问题的重要战略措施[1,2]。
煤气化是洁净煤转化的龙头技术,相较于煤燃烧,煤气化过程会释放出更高浓度的单质汞。重金属汞是有剧毒的痕量元素,单质汞与其化合物不仅会在高温下对化工设备造成腐蚀,而且对人类健康和自然环境造成极大的影响[3]。同时,单质汞相对于其他形态汞具有较高的饱和蒸气压、低水溶性和低熔点等特点,以致更难被脱除。因此,研发高效的脱除煤气中单质汞的吸附剂尤为必要。
目前,脱汞剂的制备方法有多种,各方法适用于不同的体系。赵可等[4]用浸渍法制备了铈改性半焦吸附剂(Ce/SC),考察水蒸气和α-Fe2O3对吸附剂脱汞活性的影响,结果表明水蒸气具有明显的抑制作用,但α-Fe2O3没有影响。Yang等[5]通过沉淀法制备了Fe2O3吸附剂,发现在含HCl的气氛中,100 ℃时脱汞效率可达89.5%。Qi等[6]用简易的水热合成法制备了一种脱汞性能良好且稳定的BiOlO3光催化剂。Zou等[7,8]用共沉淀法成功制备了铁钛尖晶石和铁钛锰尖晶石,发现二者在含有SO2和水蒸气等的复杂气氛中表现出了优良的脱汞性能。Zhang等[9]分别用浸渍法,溶胶-凝胶法和沉积沉淀法制备了MnOx-TiO2吸附剂,考察不同方法对吸附剂脱汞性能的影响,结果表明沉积沉淀法制备的吸附剂具有最优异的脱汞活性,这是因为活性组分MnO2有较好的氧化还原能力并且在TiO2上高度分散。在众多制备方法中,共沉淀法具有制备工艺简单,活性组分分散度高的优点。在前期工作中[10],发现1Pd5Fe/AC(活性炭负载1.0%的Pd和5.0%的Fe2O3,上述均为质量分数)双金属吸附剂在中温模拟煤气中具有优异的脱汞性能,而γ-Al2O3由于其孔结构,表面酸性以及其它性质已经被作为载体广泛用于催化领域[11,12]。因此,本工作选择了共沉淀法制备1Pd5Fe/Al2O3吸附剂,制得的吸附剂在200 ℃模拟煤气(N2-CO-H2-H2S-Hg)气氛下的固定床反应器中进行了脱汞活性评价,考察了沉淀体系pH值和焙烧温度对吸附剂的物化特性以及脱汞性能的影响,并进一步结合X射线衍射(XRD)、N2低温吸附(BET)、拉曼光谱和X射线光电子能谱(XPS)等表征结果,探讨1Pd5Fe/Al2O3吸附剂的脱汞机理。
1 实验部分
1.1 吸附剂制备
以碳酸铵(沉淀体系 pH值分别为 7.0,7.5,8.0,8.5和 9.0)作为沉淀剂,采用共沉淀法制备1Pd5Fe/Al2O3吸附剂。100 mL去离子水作为底液,一定量的PEG20000(约占固体产物质量的2%)作为分散剂,使用计量泵分别抽取一定浓度的沉淀剂溶液和硝酸铝、硝酸铁和硝酸钯的混合溶液,将两者同时匀速滴加到预先装有底液和分散剂并剧烈搅拌的烧杯中,沉淀过程中使用pH计在线测量溶液的pH值,并通过调节计量泵的流量以控制沉淀体系pH值为某一定值(误差控制在±0.05范围内),待沉淀完成后,用硝酸或氨水微调pH值至设定值,在室温下继续搅拌陈化30 min,过滤得到前驱体沉淀。然后用去离子水洗涤滤饼至近中性。将滤饼在110 ℃下干燥24 h,一定温度下焙烧5 h,即得到吸附剂,记做1Pd5Fe/Al2O3(Pd负载量1.0%,Fe2O3负载量5.0%,其余为Al2O3载体)。活性评价时选用0.25~0.42 mm(40~60目)的吸附剂颗粒。
1.2 吸附剂评价
在200 ℃模拟煤气(N2-CO-H2-H2S-Hg)气氛下的固定床反应器上对吸附剂进行了脱汞活性评价,实验装置同参考文献[10]。实验选用模拟煤气:Hg[(40±3)μg/m3]、H2S(体积分数0.02%)、H2(体积分数10%)、CO(体积分数20%)和N2载气(600 mL/min),总气体流量为1 L/min(氮气为平衡气),吸附剂的使用剂量为(500±5) mg,床层温度设定为200 ℃。样品编号FS/US-pH(x)T(y),FS和US分别代表新鲜和使用后的吸附剂,x为吸附剂制备时的pH值,y为吸附剂制备时的焙烧温度。
用吸附剂的脱汞效率(ƞ)作为模拟煤气中吸附剂脱汞性能的评价指标,其计算公式如下:

其中,C0和C1分别为反应器入口及出口的单质汞浓度,μg/m3。
1.3 吸附剂表征
吸附剂的孔结构参数采用 Micromeritics公司 ASAP2460型氮气物理吸附仪测定,利用 BET(Brunner Emmet Teller)方程计算比表面积,BJH(Barrett Joyner Halenda)方程计算平均孔径和孔容;吸附剂的XRD分析采用日本理学Rigaku Miniflex 600粉末型X射线衍射仪进行测定,角速率为8 (o)/min,步长为0.01o,2θ为10~80o;吸附剂的拉曼图谱通过英国Renishaw公司InVia型拉曼光谱仪在室温下进行测定,扫描范围100~1 500 cm-1,分辨率为1 cm-1;吸附剂的表面元素价态及含量采用英国VG公司Escalad-250型多功能X射线光电子能谱进行分析。
2 结果与讨论
2.1 pH值及焙烧温度对吸附剂的影响
沉淀体系 pH值是影响中间产物碳酸铝铵生成的关键因素。由林元华等[13]研究的 Al(NO3)3和(NH4)2CO3溶液的pH值与溶液中各物种的浓度关系可确定pH值的考察范围为7~9。焙烧温度影响晶体的生长、孔结构的形成以及活性组分的分散等,因此,焙烧温度考察范围选取500,600,700和800 ℃,并采用氮吸附和XRD等手段对所得样品进行表征,考察pH值以及焙烧温度对吸附剂物化性质和脱汞性能的影响。
2.1.1 pH值及焙烧温度对吸附剂孔结构的影响
表1为不同条件下制备的吸附剂的物理特性。由表可知,随pH值的升高,吸附剂的比表面积和孔容呈增大趋势,平均孔径逐渐减小。这主要是因为pH值升高,反应生成的碳酸铝铵沉淀含量增多,焙烧时 NH4Al(OH)2CO3分解,产生的气体带来的扩孔作用增强,最终使吸附剂的比表面积增大。由表1可知,焙烧温度对吸附剂的孔结构影响较大,其比表面积随着焙烧温度的升高先增大后减小,并且在焙烧温度为600 ℃时,具有最高的比表面积。这是由于焙烧温度较低时,产物碳酸铝铵不能完全分解,扩孔作用较弱;焙烧温度较高时,碳酸铝铵分解过快,扩孔作用过强,都会导致吸附剂比表面积较小。
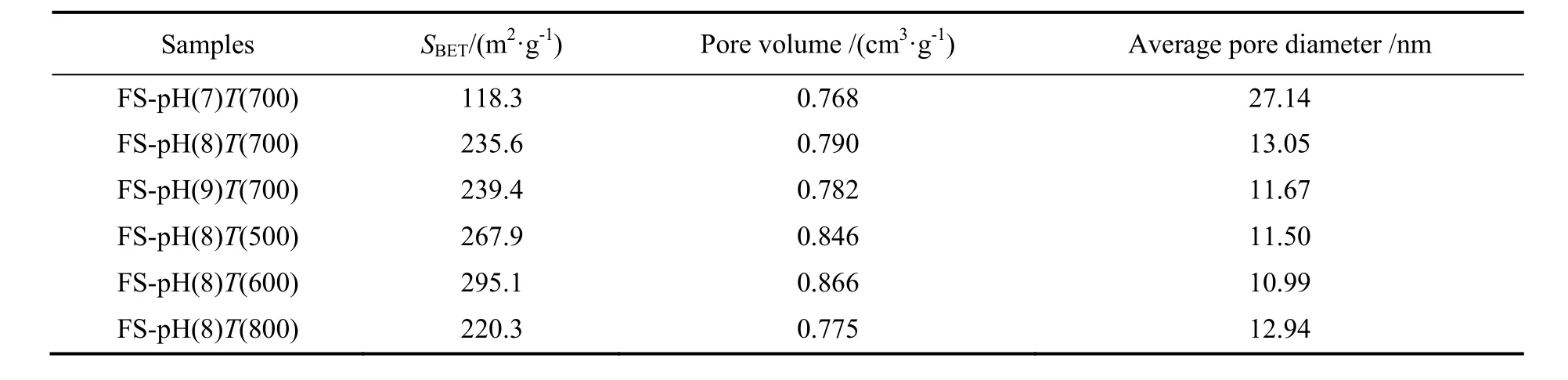
表1 不同条件下制备的吸附剂的物理特性Table 1 Physical properties of sorbents prepared under different conditions
2.1.2 pH值及焙烧温度对吸附剂晶体结构的影响

图1 不同条件下制备的样品的XRD图谱Fig.1 XRD patterns of samples prepared under different conditions
图1为不同条件下制备的吸附剂的XRD谱图。由图1(a)可知,不同pH值下制备的吸附剂在37.6,45.9和67.0°附近存在XRD衍射峰,其归属于γ-Al2O3,当在pH值为8.0,8.5和9.0时制得的吸附剂,其XRD衍射峰的强度较在pH值为7.0和7.5下制得的吸附剂的衍射峰要高,说明其结晶度更好,但峰形明显宽化,这是氧化铝晶粒细化的体现;但不同pH值下制备的吸附剂均未发现氧化钯和氧化铁的衍射峰,这是由于小晶粒衍射峰的宽化效应引起,一方面活性组分含量较少,形成的晶粒较小,另一方面活性组分在吸附剂中分散度较高。由图1(b)可知,各样品的γ-Al2O3的XRD衍射峰强度随着焙烧温度的升高,峰形逐渐变得尖锐,说明氧化铝结晶度提高,但所有样品依然未出现氧化钯和氧化铁的衍射峰,这主要是由于吸附剂中Pd和Fe相关物种含量较少且分散均匀。
2.1.3 pH值及焙烧温度对吸附剂脱汞活性的影响
图2为不同条件下制备的吸附剂的脱汞效率。由图2(a)可知,在pH值为8.0,8.5和9.0制得的吸附剂脱汞活性较高,480 min内脱汞率均可维持在90%以上;在pH值为7.0制得的吸附剂,其最初的120 min内可维持90%以上的脱汞率,但随着时间的延长,吸附剂的脱汞效率降低明显,400 min后的脱汞效率仅为20%。这是由于当制备pH值为7.0时,沉淀产物为混有勃姆石凝胶的无定型氢氧化铝,其在焙烧时团聚严重,导致吸附剂的活性组分被包藏在载体团聚体颗粒内部而无法用于脱除单质汞,另一方面是由于吸附剂FS-pH(7)T(700)的比表面积较小,活性组分在载体上的分散度较差从而导致其脱汞活性最差。
由图2(b)可知,焙烧温度为700和800 ℃时,吸附剂活性最好,480 min内吸附剂脱汞效率可保持在90%以上,但焙烧温度为800 ℃的吸附剂的脱汞活性在480 min内整体比700 ℃的略微低些;焙烧温度为500 ℃时,吸附剂活性最差,30 min后脱汞活性下降趋势剧烈,480 min时脱汞效率仅为30%;焙烧温度为600 ℃时,吸附剂的脱汞活性下降趋势缓慢,480 min时下降到71%。
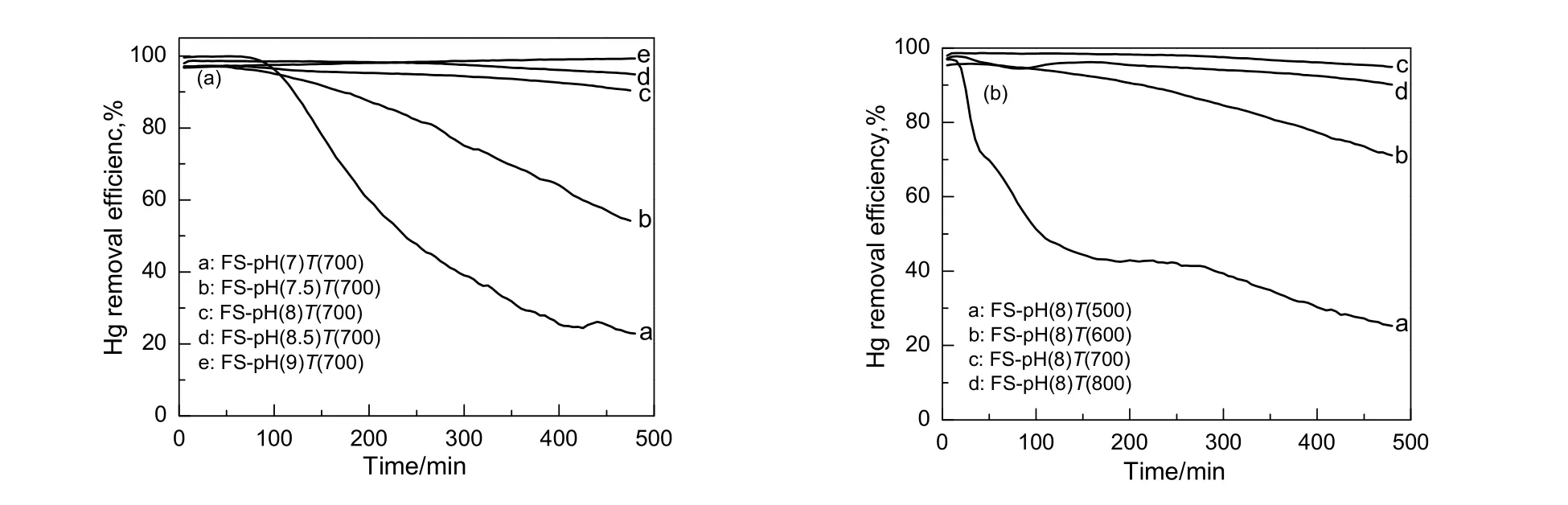
图2 不同条件下制备的1Pd5Fe/Al2O3吸附剂的脱汞效率Fig.2 Efficiency of Hg removal over the 1Pd5Fe/Al2O3 sorbents prepared under different conditions
2.2 脱汞机理探讨及制备条件对活性影响分析
2.2.1 拉曼光谱分析
图3为不同吸附剂评价前后的拉曼图谱。图中,在拉曼位移为645 cm-1处的特征峰归属于PdO,1 123 cm-1处的特征峰归属于Al2O3,1 327 cm-1处为Fe2O3的特征峰。由图可知,相同pH值下,焙烧温度越高,吸附剂在645 cm-1处PdO的特征峰越强,且出现了微弱的氧化铁的峰,这说明随着焙烧温度的升高,吸附剂中各组分晶粒变大,晶体规整度提高,这与XRD表征结果基本一致。相同焙烧温度下,吸附剂中PdO的峰强度随着pH值的升高先增强后减弱,这说明随着pH值的升高,晶体规整度与PdO分散的均匀性同时提高,而提高活性组分,晶体规整度和分散性有助于吸附剂对汞的脱除。这一结果与图2(a)所示的评价结果相吻合。脱汞活性评价后,所有吸附剂在645 cm-1处的PdO的特征峰几乎消失,可能是由于PdO直接参与了反应,或者被煤气中的还原性组分还原成了Pd0,而Pd0的含量较少,因而谱图中没有出现Pd0的拉曼特征峰。
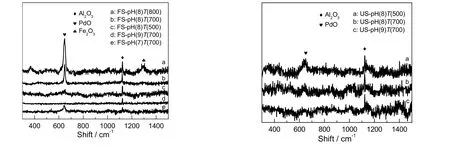
图3 吸附剂评价前后的Raman图谱Fig.3 Raman spectra of fresh and used sorbents
2.2.2 X射线光电子能谱分析
为了揭示吸附剂的脱汞活性组分化学信息,对评价前后的pH(8)T(700)吸附剂进行了XPS分析,结果如图4所示。
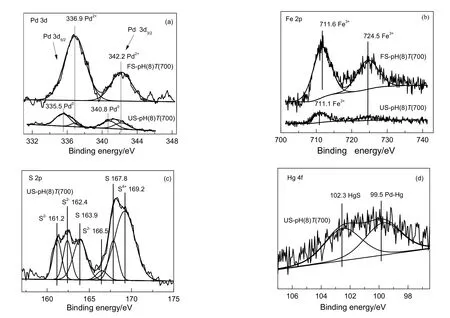
图4 吸附剂评价前后的XPS图谱Fig.4 XPS spectra of fresh and used sorbents
由图4(a)可知,对于新鲜吸附剂,PdO的3d5/2和3d3/2的结合能分别为336.9和342.2 eV[14,15];活性评价后的吸附剂上除了存在Pd2+,还含有结合能位于335.5和 340.8 eV的Pd0[16]。研究表明[17],PdO的还原温度在60 ℃左右,即在活性评价温度200 ℃下PdO被H2和CO迅速还原为Pd0。由图4(b)可知,评价后Fe2O3的峰强度变弱,这可能是由于Fe2O3参与了活性评价过程中的某些化学反应[10]。由图4(c)可知,在161.2,162.4和163.9 eV处有3个硫元素的结合能,其分别可归属为PdS[18],FeS[19]以及单质S[20]。由图4(d)可知,Hg元素存在99.5和102.3 eV[21]两个结合能,其分别归属于Hg0和Hg2+,其对应的物质为Pd-Hg合金及HgS[22]。
XPS能谱图中光电子谱线强度(光电子峰的面积)可以反映吸附剂表面原子的含量。不同吸附剂评价前后Pd,Fe和S的表面含量如表2所示。对于新鲜吸附剂,500,700和800 ℃下焙烧的吸附剂上PdO表面含量分别为0.06%,0.09%及0.10%,氧化铁的表面含量依次为0.96%,0.99%及1.11%,吸附剂评价后表面硫含量依次1.11%,1.99%及2.22%。结果表明,随着焙烧温度的升高,Pd和Fe的表面元素含量增高,说明较高温度的焙烧能使吸附剂中的PdO和Fe2O3发生迁移,由吸附剂的体相向表面迁移聚集,但温度过高又会使活性组分团聚,最终导致吸附剂的活性下降,与活性评价结果一致。活性评价后Fe2O3的表面含量明显降低,这是因为Fe2O3可以与煤气中的H2S反应而被消耗;表面的硫含量随着Fe含量的增加而增加,是由于含S物种(单质硫,FeSx,PdS和HgS等)的生成。

表2 吸附剂评价前后Pd,Fe和S的表面含量Table 2 Contents of Pd, Fe and S on the surface of fresh and used sorbents
通过上述表征结果可以得到Pd-Fe/Al2O3吸附剂的脱汞机理如下所示:

3 结 论
以硝酸铝、硝酸铁和硝酸钯为前驱体溶液,以碳酸铵为沉淀剂,采用共沉淀法成功制备了双金属吸附剂1Pd5Fe/Al2O3。实验表明沉淀体系pH值和焙烧温度对吸附剂的物理结构及化学性质有较大影响。当pH值大于8时可生成碳酸铝铵沉淀,且生成量随pH值升高而增加,碳酸铝铵在焙烧时可分解产生NH3,CO2和H2O等气体带来扩孔作用。吸附剂的比表面积随焙烧温度的升高先增大后减小,这是由于焙烧温度过低时,碳酸铝铵分解不充分使扩孔作用较弱,焙烧温度过高时,碳酸铝铵分解过快使扩孔作用过强,均会导致吸附剂比表面积变小。在pH值为8,焙烧温度为700 ℃的条件下制得1Pd5Fe/Al2O3具有较高的比表面积,且活性组分Pd表面含量最高、分散较为均匀,该条件下制备的吸附剂脱汞效率在480 min内可维持在95%以上,具有较佳的脱汞活性。
[1]徐振刚. 煤气化推动中国洁净煤技术发展[J]. 中国煤炭, 2007, 33(7): 8-9.Xu Zhengang. Coal gasification promotes CCT development in China[J]. China Coal, 2007, 33(7): 8-9.
[2]倪维斗, 李 政. 以煤气化为核心的多联产能源系统—资源/能源/环境整体优化与可持续发展[J]. 煤化工, 2003, 31(1): 3-10.Ni Weidou, Li Zheng. Multi-generation energy system from coal gasification process[J]. Coal Chemical Industry, 2003, 31(1): 3-10.
[3]Johari K, Saman N, Song S T, et al. Utilization of coconut milk processing waste as a low-cost mercury sorbent[J]. Industrial &Engineering Chemistry Research, 2013, 52(44): 15648-15657.
[4]赵 可, 牛庆欣, 王 力, 等. 水蒸气和α-Fe2O3对铈改性半焦脱除单质汞的影响研究[J]. 燃料化学学报, 2017, 45(3): 378-384.Zhao Ke, Niu Qingxin, Wang Li, et al. Effect of water vapor andα-Fe2O3on elemental mercury removal performance over cerium oxide modified semi coke[J]. Journal of Fuel Chemistry and Technology, 2017, 45(3): 378-384.
[5]Yang Y, Liu J, Wang Z, et al. Heterogeneous reaction kinetics of mercury oxidation by HCl overα-Fe2O3surface[J]. Fuel Processing Technology, 2017, 159: 266-271.
[6]Qi X M, Gu M L, Zhu X Y, et al. Fabrication of BiOIO3nanosheets with remarkable photocatalytic oxidation removal for gaseous elemental mercury[J]. Chemical Engineering Journal, 2016, 285: 11-19.
[7]Zou S, Liao Y, Xiong S, et al. H2S-modified Fe-Ti spinel: a recyclable magnetic sorbent for recovering gaseous elemental mercury from flue gas as a Co-benefit of wet electrostatic precipitators[J]. Environmental Science & Technology, 2017, 51(6): 3426-3434.
[8]Xiong S, Xin X, Nan H, et al. Elemental mercury oxidation over Fe-Ti-Mn spinel: performance, mechanism and reaction kinetics[J].Environmental Science & Technology, 2017, 51(1): 531.
[9]Zhang A C, Zhang Z H, Shi J M, et al. Effect of preparation methods on the performance of MnOx-TiO2adsorbents for Hg0removal and SO2resistance[J]. Journal of Fuel Chemistry and Technology, 2015, 43(10): 1258-1266.
[10]Han L, He X, Yue C, et al. Fe doping Pd/AC sorbent efficiently improving the Hg0removal from the coal-derived fuel gas[J]. Fuel, 2016,182: 64-72.
[11]Potdar H S, Jun K W, Bae J W, et al. Synthesis of nano-sized porous γ-alumina powder via a precipitation/digestion route[J]. Applied Catalysis A General, 2007, 321(2): 109-116.
[12]Parida K M, Pradhan A C, Das J, et al. Synthesis and characterization of nano-sized porous gamma-alumina by control precipitation method[J]. Materials Chemistry & Physics, 2009, 113(1): 244-248.
[13]林元华, 张中太, 黄传勇, 等. 前驱体热解法制备高纯超细α-Al2O3粉体[J]. 硅酸盐学报, 2000, 28(3): 268-271.Lin Yuanhua, Zhang Zhongtai, Huang Chuangyong, et al. Preparation of high purity and ultrafineα-Al2O3powders by pyrolysis of NH4AlO(OH)HCO3[J]. Journal of The Chinese Ceramic Society, 2000, 28(3): 268-271.
[14]Voogt E H, Mens A J M, Gijzeman O L J, et al. XPS analysis of palladium oxide layers and particles[J]. Surface Science, 1996, 350(1/3):21-31.
[15]Brun M, Berthet A, Bertolini J C. XPS, AES and auger parameter of Pd and PdO[J]. Journal of Electron Spectroscopy & Related Phenomena, 1999, 104(1/3): 55-60.
[16]Datye A K, Bravo J, Nelson T R, et al. Catalyst microstructure and methane oxidation reactivity during the Pd↔PdO transformation on alumina supports[J]. Applied Catalysis A General, 2000, 198(1/2): 179-196.
[17]Hou W, Zhou J, Yu C, et al. Pd/Al2O3sorbents for elemental mercury capture at high temperatures in syngas[J]. Indengchemres, 2014,53(23): 9909-9914.
[18]Liu S, Wang X, Wang K, et al. ZnO/ZnS-PdS core/shell nanorods: synthesis, characterization and application for photocatalytic hydrogen production from a glycerol/water solution[J]. Applied Surface Science, 2013, 283(11): 732-739.
[19]Chen H, Zhang Z, Yang Z, et al. Heterogeneous fenton-like catalytic degradation of 2,4-dichlorophenoxyacetic acid in water with FeS[J].Chemical Engineering Journal, 2015, 273: 481-489.
[20]Zhang H, Zhao J, Fang Y, et al. Catalytic oxidation and stabilized adsorption of elemental mercury from coal-derived fuel gas[J]. Energy Fuels, 2012, 26(3): 1629-1637.
[21]Hutson N D, Attwood B C, Scheckel K G. XAS and XPS characterization of mercury binding on brominated activated carbon[J].Environmental Science & Technology, 2007, 41(5): 1747-1752.
[22]Hyland M M, Jean G E, Bancroft G M. XPS and AES studies of Hg(II) sorption and desorption reactions on sulphide minerals[J].Geochimica et Cosmochimica Acta, 1990, 54(7): 1957-1967.
