盐酸决奈达隆的合成工艺改进
2018-01-12邱江凯王德才
王 晶 邱江凯 王德才
(南京市第一中学,江苏 南京 210001;*南京工业大学,江苏 南京 211800)
盐酸决奈达隆(Dronedarone hydrochloride,1)是赛诺菲-安万特用了近20年时间研发成功的一种新的抗心律失常药物,2009年7月经美国食品药品监督管理局(FDA)批准上市,适用于患有阵发性或持续性房颤或房扑患者。其化学结构与治疗房颤的标准药物胺碘酮相似,作用机制相同,均为钾离子通道阻滞剂。但在侧链苯环的第 3和第5位上无碘原子,故无甲状腺和肺毒性,是一种新型的、更安全有效的治疗心律失常的药物;盐酸决奈达隆是目前经临床试验证明唯一显示出能够显著降低房颤/心房扑动患者发病率和死亡率的抗心律失常药物,是过去20年里,抗心律失常药物研发领域里具里程碑意义的创新新药。因为此前还没有一个药物被证明能够降低心律失常患者的死亡率。盐酸决奈达隆是第一个在美国被批准上市的房颤和房扑治疗新药。是治疗房颤和房扑使之迅速转复为窦性心律的最佳药物疗法,因此是一种理想的治疗心血管病药物,具有广阔的临床应用前景[1~3]。
文献报道盐酸决奈达隆的合成方法较多[1,4~11],分别主要是以2-正丁基-3-(4-羟基苯甲酰基)-5-硝基苯并呋喃(2)[4,5]、2-正丁基-5-硝基苯并呋喃[6,7]或2-正丁基-5-氨基苯并呋喃[8]为关键中间体经3~5步反应制得盐酸决奈达隆;在这些合成路线中以2为关键原料的合成方法较有工业化应用前景[13],文献[13]报道以2为起始原料,与1-溴-3-氯丙烷进行醚化反应得到2-正丁基3-[4-(3-氯丙氧基)苯甲酰基]-5-硝基苯并呋喃(3),再利用Fe-NH4Cl将其还原成2-正丁基-3-[4-(3-氯丙氧基)苯甲酰基]-5-胺基苯并呋喃(4),再与甲基磺酰氯进行甲磺酰胺化反应得到2-正丁基-3-[4-(3-氯丙氧基)苯甲酰基]-5-甲磺酰胺基苯并呋喃(5),然后将之与二正丁胺进行N-烃化反应得到决奈达隆(6),对6用氯化氢进行成盐反应,得到1。该路线具有原料易得且便宜,工艺较稳定,反应总收率较高等特点,但还存在可能产生二甲磺酰胺化副产物和铁泥污染及需使用氯化氢气体等弊端。
[4~14]报道路线并作合理改进,以2为起始原料,与1-溴-3-氯丙烷进行醚化反应得到3;再利用Pd/C催化氢化将3还原成4;4与甲基磺酰氯进行甲磺酰胺化反应得到5;然后将5与二正丁胺进行N-烃化反应得到6;6用盐酸进行成盐反应,得到1, 该工艺参考文献[13,14]报道路线并作合理改进,整个反应原料易得且便宜,反应条件温和,操作方便,工艺稳定,制得的产品纯度高,易于工业化生产。合成路线如图1所示:
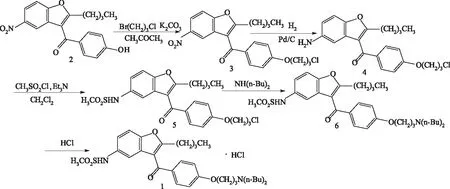
图1 盐酸决奈达隆合成路线Fig 1 Synthetic route of Dronedarone hydrochloride
1 实 验
1.1 试剂
2-丁基-3-(4-羟基苯甲酰基)-5-硝基苯并呋喃(工业级,纯度99.45%),南京康满林生物医药科技有限公司;钯碳(5%,工业级),陕西瑞科新材料股份有限公司;1-溴-3-氯丙烷、丙酮、碳酸钾、正己烷、乙酸乙酯、甲苯、乙醇、二氯甲烷、三乙胺、甲基磺酰氯、二正丁胺、浓盐酸等均为化学纯。
1.2 仪器
X4显微熔点仪(温度计未经校正);AV-500型核磁共振仪;TOF-MS质谱仪。
1.3 实验步骤
1.3.12-正丁基-3-[4-(3-氯丙氧基) 苯甲酰基]-5-硝基苯并呋喃(3)的制备[13,14]
向500 mL反应瓶中加入200 mL丙酮、33.9 g (0.100 mol)2、15.2 g(0.110 mol)无水碳酸钾、23.6 g (0.150)1-溴-3-氯丙烷,搅拌并升温至回流反应10 h, TLC跟踪至原料反应完全。结束反应,将反应液冷却至30℃,过滤,用30 mL丙酮淋洗滤饼3次,洗涤液、滤液合并,减压蒸馏至无馏分出现。降温至30~40 ℃,加入100 mL正己烷,同时搅拌直至析出晶体,降温至0~5 ℃,析出大量晶体。过滤,用20 mL正己烷淋洗滤饼,抽干,真空干燥得3,重39.8 g,收率95.7%。
1.3.22-正丁基-3-[4-(3-氯丙氧基)苯甲酰基]-5-胺基苯并呋喃(4)的制备
向500 mL氢化反应釜中加入39.5 g(0.095 0 mol)3溶于120 mL乙酸乙酯的溶液,加入3.95 g 5%钯炭(湿重),再加入40 mL的乙酸乙酯,于氢压约0.20 Mpa、温度约50 ℃,搅拌反应3 h, TLC跟踪至原料反应完全。结束反应,过滤,洗、滤液合并。水浴减压蒸馏至基本无馏分。加入30 mL甲苯和120 mL正己烷,冷却,搅拌析晶。内温降至0~10℃,析出大量固体,过滤,用30mL正己烷淋洗滤饼,抽干,真空干燥得4,重36.3 g,收率99.0%。
1.3.32-正丁基-3-[4-(3-氯丙氧基) 苯甲酰基]-5-甲磺酰胺基苯并呋喃(5)的制备[13]
在500 mL反应瓶中加入35.9 g(0.093 0 mol)4和150 mL二氯甲烷,冷却至-10~-5 ℃。加入12.8 g(0.112 mol)甲磺酰氯,控制温度在-10~0 ℃,缓慢滴加10.3 g(0.102 mol)三乙胺于100 mL二氯甲烷的溶液,TLC跟踪至原料反应完全。加入100 mL水,搅拌,并升温至室温,静置,分液;有机层加入20 g无水硫酸钠干燥,过滤,滤液减压蒸馏至无馏分出现。加入50mL乙酸乙酯,加热温度升至75~80 ℃,浓缩液全部溶解,滴加正己烷约100 mL,停止加热,降至室温,再缓慢降至0~5 ℃,搅拌析晶,抽滤,滤饼用乙酸乙酯∶正己烷=1∶4的混合溶剂20 mL进行洗涤,抽干,45~50 ℃真空干燥箱干燥8 h,得灰白色固体5,重37.5 g,收率86.9%。
1.3.4盐酸决奈达隆(1)的制备
在500 mL反应瓶中加入37.1 g(0.080 mol)5,加入62.0 g(0.480)二正丁胺。搅拌下加热溶解并升温至100 ℃,保温反应约6 h后, TLC跟踪至原料反应完全,停止加热。冷却至室温,加入100 mL水。加入150 mL二氯甲烷,搅拌5 min,静置、分液,有机层加入15 g硅胶(300~400目),搅拌至少1 h。过滤,用约50 mL二氯甲烷淋洗滤饼,合并洗、滤液,将其转移至1 000 mL的反应瓶中,搅拌,将内温降至-10~-5 ℃。控制温度-10~0 ℃,缓慢滴加2 mol/L稀盐酸约250 mL,滴加完毕后,控制温度-10~0 ℃,搅拌反应1 h。静置,分液,有机层每次用150 mL水洗3次,有机层加入30 g无水硫酸钠搅拌干燥2 h。过滤,滤液减压蒸至基本无馏分。加入70 mL二氯甲烷,加热、搅拌至浓缩液溶清,再加入180 mL丙酮,冷却,内温降至0~5 ℃,搅拌析晶。继续冷却析晶1 h,过滤,用60 mL丙酮分次淋洗滤饼,抽干,经真空干燥得白色固体1,熔点142.5~143 ℃(文献[4]143℃),重40.7 g,收率85.8%。经HPLC检测纯度为99.85%。
2 结果与讨论
2.1 终产物盐酸决奈达隆的结构确证
2.1.1核磁共振图谱解析
1H NMR (500 MHz, DMSO-D6)δ: 11.00 (br,s、1H) ,9.66 (s、1H) , 7.80(d、2H、J=8.5),7.62(d、1H、J=9.0),7.31(d、1H、J=2.0),7.25(dd、1H、J=9.0、2.0),7.11(d、2H、J=8.5),4.24(t、2H、J=6.0),3.23(m、2H),3.06(m、4H),2.90(s、3H),2.82(t、2H、J=7.5),2.22~2.27(m、2H),1.63~1.74(m、6H),1.34(六重峰、4H、J=7.5),1.25(六重峰、2H、J=7.5),0.92(t、6H、J=7.5),0.81(t、3H、J=7.5)。13CNMR(125MHz,DMSO-D6)δ:189.11,164.52,162.11,150.25,134.22,131.28,131.08,127.15,118.81,116.30,114.41,113.20,111.48,65.27,51.58,48.66,38.54,29.32,27.09,24.74,22.76,21.51,19.39,13.41,13.28。
2.1.2质谱解析
用TOF-MS质谱仪测定产物的高分辨ESI+图谱,样品的高分辨质谱图显示其准分子离子峰为557.3063,为(M-HCl+H)+峰,对应的分子式为C31H45N2O5S,与决奈达隆(C31H44N2O5S)分子式吻合,说明样品的分子式为C31H45ClN2O5S,这与盐酸决奈达隆的分子式吻合。
2.2 讨论
第一步反应采用将起始原料2与1-溴-3-氯丙烷、缚酸剂一锅法混合反应,与已有工艺相比较缩短了反应时间,简化了反应步骤,反应的析晶方式上采用一种独特的析晶方式,使析出的晶体,粒径均一,避免了原工艺析晶时不规则块状固体产生的现象,有利于工业化生产放大。第二步反应原工艺用铁粉还原硝基不仅需要高温,消耗大量的金属铁,而且产生的大量铁泥极易污染环境,不适合工业化生产,所以本项目采取了新的金属催化氢化的还原方法,这种方法较铁粉还原硝基来说更加清洁环保,产物的收率更高,纯度也更好,并且使用的金属钯也能多次回收利用,大大节省了成本。第三步甲基磺酰化反应上采用新的投料方式,十分有效地控制了甲磺酰胺化双取代杂质的生成,使甲磺酰基双取代的杂质含量控制在1.0%以下,大大提高了产物的纯度;第四步N-烃化反应按照文献的工艺描述制得的决奈达隆(游离碱)是必须通过硅胶柱层析的方法才能得到的,且收率偏低,不适合工业化生产。现有工艺在胺化反应的后处理上采取了大胆的创新,就是N-烃化反应后不需纯化,而是直接与盐酸进行成盐反应得到决奈达隆盐酸盐,成盐后再采取一些简单的处理方法得到盐酸决奈达隆粗品,经进一步精制得到盐酸决奈达隆纯品。
3 结 论
改进后的工艺起始原料廉价易得,反应条件温和,操作简便,绿色环保,制得的产品纯度高,总收率可达70.6%,适合于工业化放大。
参考文献
[1] 何晓清,吴泰志,张福利等.盐酸决奈达隆合成路线图解[J].中国医药工志,2010,02:148~151.
[2] Patel P D,Bhuriya R,Patel D D.,et al. Dronedarone for atrial fibrillation: a new therapeutic agent[J].Vasc Health Risk Manag,2009 (5) : 635~642.
[3] Connolly S J,Crijns H J,Torp-Pedersen C.,et al.Analysis ofstroke in ATHENA: a placebo-controlled, double-blind, parallel-arm trial to assess the efficacy of dronedarone 400 mg BID for theprevention of cardiovascular hospitalization or death from anycause in patients with atrial fibrillation / atrial flutter[J].Circulation, 2009,120: 1 174~1 180.
[4] Gubin J,Lucchetti J,Inion H.,et al.Alkylaminoalkyl derivatives of benzofuran,benzothiophene ,indolizine, process for their preparation and compositions containing them[P].US5223510A,1993-06-29.
[5] Eklund L,Cambrex K. Process for preparing benzofurans[P].WO 2009044143A2,2009-04-09.
[6] M.比尔德.苯甲酰基衍生物和其盐酸盐及其制备方法 [P].中国1769262A,2006-05-10.
[7] N.诺菲,C.勒鲁瓦.甲基磺酰胺基——苯并呋喃衍生物,它的制备方法与它作为合成中间体的应用[P].中国1479735A,2004-03-03.
[8] Arie G,Gennady N,Lev Y. Process for the preparation of dronedarone[P].WO 03/ 04120A1.2004-05-15.
[9] 汪玉梅,张勇,胡永亮. 盐酸决奈达隆的合成[J]. 河北化工,2012,35(4):12~13.
[10] 王冠,张飞龙,王佳静 等.盐酸决奈达隆的合成[J]. 中国医药工业杂志,2011,42(12):881~883.
[11] 李峰,田拴红,宋晓峰 等. 盐酸决奈达隆合成新工艺[J]. 高等学校化学学报,2013,34(4):858~862.
[12] 李素义,钟启星,陈国华 等.盐酸决奈达隆的合成[J]. 中国医药工业杂志,2011,42(3):161~164.
[13] 何学军,宋俊松,王德才 等.盐酸决奈达隆的合成[J]. 生物加工过程,2012,10(3):67~70.
[14] 王德才,宋俊松,刘华权 等.一种盐酸决奈达隆的制备方法[P].中国102382087A, 2012-03-21.
