基于二维最大重叠离散小波变换的代谢组质谱数据的预处理方法*
2018-01-03李贞子
邓 魁 李贞子 侯 艳 李 康
哈尔滨医科大学卫生统计学教研室(150081)
·论著·
基于二维最大重叠离散小波变换的代谢组质谱数据的预处理方法*
邓 魁 李贞子 侯 艳 李 康△
哈尔滨医科大学卫生统计学教研室(150081)
目的通过二维最大重叠离散小波变换(maximal overlap discrete wavelet transform,MODWT)对代谢组学质谱数据进行预处理,去除一定的噪声和批次效应,提高分析方法的有效性和稳定性。方法针对卵巢癌和卵巢囊肿的质谱数据,选取Haar小波函数对其进行二维MODWT的数据变换,获得不同尺度的数据,再对其中的细节数据置0和进行重构。对预处理后的数据用随机森林(RF)方法筛选差异变量和建立判别模型,评价预处理的效果。结果经过二维MODWT处理后的质谱数据建模判别效果明显优于使用原始数据得到的结果。结论针对质谱数据,二维MODWT方法能够更好地进行特征提取,提高模型的判别能力,具有研究价值和应用价值。
代谢组学 质谱数据 数据预处理 小波变换
代谢组质谱数据是指使用超高效液相色谱-质谱联用仪检测血液、尿液或组织等生物样品得到的数据,目的是衡量不同的内源小分子的相对含量[1]。由于检测结果受样品的预处理、环境温度、色谱柱效的改变等多种因素影响,数据中可能含有大量的噪声和一定的批次效应,使检测得到的数据极不稳定,从而影响数据的分析结果[2]。
与基因组学和蛋白质组学数据相比,代谢组学数据由于代谢物的结构具有更大的差异性,对其进行标准化有更大的难度,目前尚无一种标准的公认方法对代谢组学数据进行标准化预处理[3]。针对代谢组学数据的预处理,现在常见的统计学方法有Z标准化、最大峰归一化和总和峰归一化等[4]。Z标准化能够消除不同代谢物浓度数量级的差别,但无法去除噪声的干扰。最大峰归一化和总和峰归一化能够消除某些混杂因素对代谢物浓度的干扰,其局限性在于它们假定在机体紊乱时,一组代谢物浓度的上升同时伴随着另一组代谢物浓度的下降,而这种假定在实际中往往并不成立[5]。本文给出一种新的基于二维最大重叠Haar小波变换的数据预处理方法,这种方法利用了代谢组学质谱数据时间序列的特性,将数据分解成不同的特征,从而能够较好地去除由于不稳定因素引起的数据波动,提高判别分析的效果。
原理与方法
代谢组质谱数据可记为D={xij}(i=1,2,…,n;j=1,2,…,m),其中n和m分别表示检测的样本数和物质(变量)数目。
二维最大重叠离散小波变换(maximal overlap discrete wavelet transform,MODWT)是从二维离散小波变化改进得来的,但又与传统的二维离散小波变化有所不同[6]。二维离散小波变换的分解和重构过程如图1所示,即首先对原始数据的每一行样品进行小波变换,获得低频分量L和高频分量H;然后再进一步作小波变换的列变换,得到行列都为低频分量(LL1)、行为低频分量列为高频分量(LH1)、行为高频分量列为低频分量(HL1)、行列皆为高频分量(HH1)四部分数据。第二次数据变换可以在LL1上继续进行得到LL2、LH2、HL2和HH2。以此类推可以变换多次。其中低频分量在一定程度上反映了数据本质信息,本质特征。而高频分量则在一定程度上反映了噪声信息,如在实验过程中环境温度、色谱柱效的改变等多种系统误差的干扰以及随机误差的影响。最后得到的变换可以通过逆运算,完全重构回原始数据。实际中,可以对分解后的数据进行适当处理,如对某部分数据置0或按一定阈值置0后进行重构,从而达到去除无用的检测信号的目的。
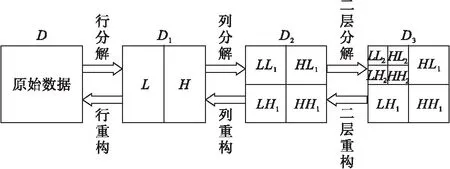
图1 二维离散小波变换分解和重构过程示意图
上述小波行变换的公式为:
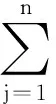
(1)
(2)
(3)
其中i表示相应的行,k表示变换后相应列的数据,L(i,k)表示变换后的低频分量,H(i,k)表示变换后的高频分量。h(·)为尺度函数,g(·)为小波函数。
进一步,对L和H作如下分解:
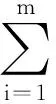
(4)
(5)
(6)
(7)
(8)
同理可以得到D3。上式中LL(k,j)表示对L作小波列变换得到的低频分量,LH(k,j)表示对L作小波列变换得到的高频分量,HL(k,j)表示对H作小波列变换得到的低频分量,HH(k,j)表示对H作小波列变换得到的高频分量。h(·)为尺度函数,g(·)为小波函数。
本文使用Haar小波,其尺度函数为:

(9)
Haar小波函数定义为尺度函数的对偶函数,即g(x):=h(2x)-h(2x-1)。这是一个阶跃函数,它具有非连续性,具有在时域区间内局部化能力,其函数为

(10)
二维MODWT更适合代谢组学数据分析,因为它允许小波系数和原始数据变量相对应。根据小波变换理论,最有意义的信息包含在较大的系数中,噪声一般在较小的系数中,因此,仅重构较大的系数就可以保持原始数据的本质特征。
本研究用上述二维最大重叠离散小波变换对质谱数据进行预处理,然后用随机森林(random forest,RF)模型进行判别分析。
实例分析
实例:2011年3月至2013年7月从哈尔滨医科大学附属肿瘤医院妇科收集了140例上皮性卵巢癌(EOC)患者和158例卵巢囊肿(BOT)患者血浆样品。得到超高效液相色谱质谱联用检测结果。原始数据通过MASS-Hunter定性分析软件转化为mzdata格式的文件,这些文件导入到R中通过XCMS包进行预处理。最后得到2106个变量。
用二维MODWT对原始数据分别进行一层和两层分解,得到D2和D3(参见图1),其中HH1和HH2部分主要包含细节噪声信息,对其进行置0处理,然后对数据重构。为了验证预处理的效果,在140例EOC患者中随机抽取70例患者,158例BOT患者中随机抽取79例患者作为训练集,其余样品作为测试集。在训练集中使用RF方法筛选出重要性评分在前50位的特征建立判别模型,对测试集样品进行分类和预测,通过真实分类标签计算ROC曲线下的面积AUC值。上述试验通过无重复抽样进行1000次,最后通过1000个AUC的平均值考核方法的有效性。小波变换和RF判别模型均通过R语言程序包实现。
试验结果:经RF判别后得到AUC值后的结果如图2、图3和图4所示。由图看出,代谢组质谱原始数据进行RF判别分析后得到AUC值为0.87±0.03,经两种不同变换再进行RF判别得到AUC值分别为0.94±0.02和0.96±0.02。图2和图3给出了Haar小波变换与原始数据的AUC值频数图,结果显示经Haar小波变换后,卵巢癌质谱数据的分类能力有了很大的提高,并且随着分解层数从第一层增加到第二层,分类能力进一步提高。图4给出了原始数据,一层小波变换后数据,两层小波变换后数据分别经RF判别得到的判别结果的ROC曲线图,结果显示二维MODWT能明显提高RF的分类能力。
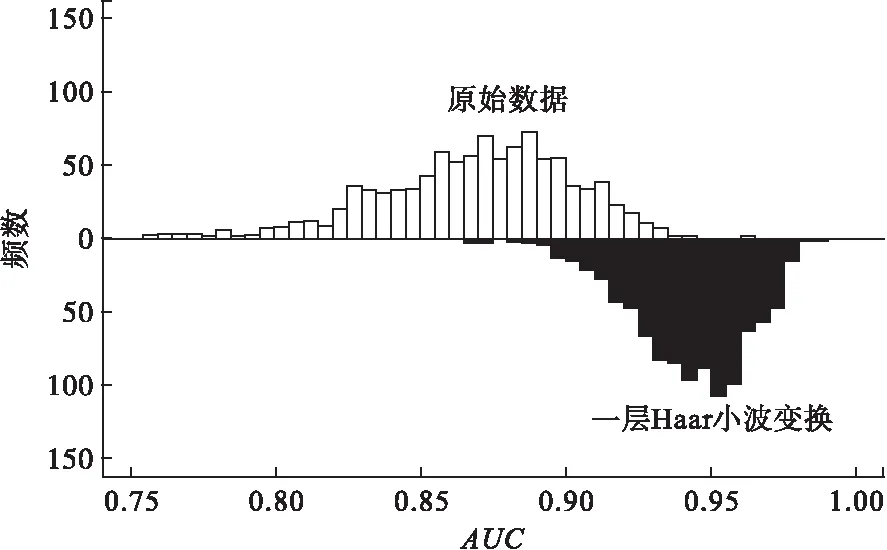
图2 一层Haar小波变换后AUC值频数变化情况
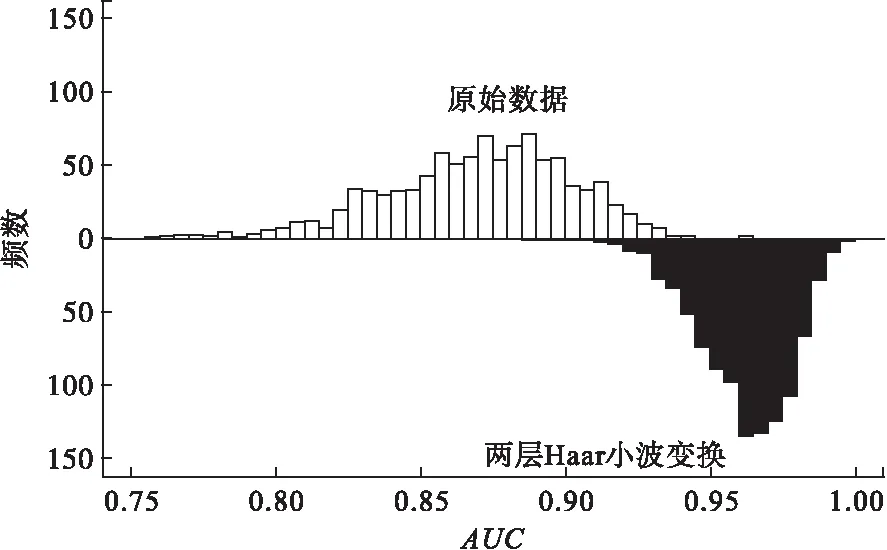
图3 两层Haar小波变换后AUC值频数变化情况
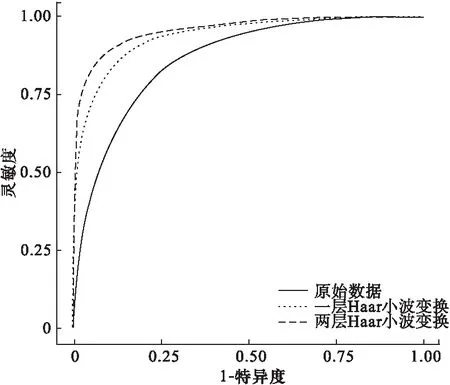
图4 原始数据和两种不同层小波变换的RF模型的ROC曲线
讨 论
本文尝试将二维小波变换应用于代谢组学数据预处理,其基本思想是利用质谱相邻间物质具有一定的相关性、检测样品间时间上的相关性,以及同类观测样品(如癌症和非癌)的聚类性质,进行二维小波变换,适当去除干扰信号,获得更好的分类效果。通过小波变换后,RF模型的判别效果得到显著的提高。
如果将本文的实例通过随机打乱样品标签后再进行二维MODWT处理,然后进行RF判别后得到的AUC值均在0.5附近,这进一步证明了本研究得到结果的可信度。
在之前的研究中,我们使用常用的Z标准化、最大峰归一化和总峰和归一化的方法对代谢组学数据进行预处理,发现RF的判别能力并不能得到提升,其AUC值分别为0.87±0.03、0.87±0.03,0.85±0.03。
不同的小波函数也有不同的特点。因此在二维离散小波变换中,需要选择合适的小波函数。研究表明,Haar小波函数对代谢组质谱峰数据进行质谱峰提取能够获得更好的结果[8],因此本文选用Haar小波。然而,实际中究竟选用何种小波函数好,与数据结构和研究目的有关,对此需要进一步研究。
本研究中分解层数为一层和二层时,达到了较好的预测能力。实际我们在进行更多层次处理后发现,分类能力虽然越来越强,甚至接近1,但随机打乱分类标签再进行处理后,RF的判别效果依然很高,甚至接近0.9,这说明这种分类能力提高是虚假的提高,可能出现过拟合的现象。在实际研究中,要根据数据结构和特点选择最佳的分解层数。
本研究通过二维最大重叠离散小波变换,将数据分解为行列都为低频分量(LL)、行为低频分量列为高频分量(LH)、行为高频分量列为低频分量(HL)、行列皆为高频分量(HH)四部分数据,考虑到细节系数(HH)主要包含噪声信息[10],因此将其置0处理,再进行重构,从而达到去除一定的随机波动,保留原始数据主要特征信息的目的,这只是一种简单的处理方法。更好的方法应该根据实际数据,对行为低频分量列为高频分量(LH)、行为高频分量列为低频分量(HL)这两部分数据有选择地使用硬阈值[11]或软阈值[12]的方法进行处理,既能去除干扰信号,又能够保留有效的检测数据信息。对此有待于更进一步的研究。
[1] Wang Y,Hu H,Su Y,et al.Potential of monitoring isotopologues by quantitative gas chromatography with time-of-flight mass spectrometry for metabolomic assay.Journal of Separation Science,2016,39(6):1137-1143.
[2] De Livera AM,Sysi-Aho M,Jacob L,et al.Statistical methods for handling unwanted variation in metabolomics data.Analytical Chemistry,2015,87(7):3606-3621.
[3] Wu Y,Li L.Sample normalization methods in quantitative metabolomics.Journal of Chromatography A,2016,1430:80-95.
[4] 柯朝甫,张涛,武晓岩,等.代谢组学数据分析的统计学方法.中国卫生统计,2014,31(2):357-359.
[5] Sysi-Aho M,Katajamaa M,Yetukuri L,et al.Normalization method for metabolomics data using optimal selection of multiple internal standards.BMC Bioinformatics,2007,8(1):1-17.
[6] P Liò.Wavelets in bioinformatics and computational biology:state of art and perspectives.Bioinformatics,2003,19(1):2-9.
[7] 彭娟,李川.基于最大重叠离散小波变换的油中颗粒污染物特征信号提取.重庆工商大学学报(自然科学版),2013,30(6):24-28.
[8] Davis RA,Chariton AJ,Godward J,et al.Adaptive binning:An improved binning method for metabolomics data using the undecimated wavelet transform.Chemometrics & Intelligent Laboratory Systems,2007.85(1):144-154.
[9] Yang Y,He Y,Cheng J,et al.A gear fault diagnosis using Hilbert spectrum based on MODWPT and a comparison with EMD approach.Measurement,2009,42(4):542-551.
[10]Lewis AS,Knowles G.Image compression using the 2-D wavelet transform.IEEE Transactions on Image Processing,1992,1(2):244-50.
[11]Chen C,Zhou N.A new wavelet hard threshold to process image with strong Gaussian Noise.IEEE Fifth International Conference on Advanced Computational Intelligence,2012:558-561.
[12]Fang Y.A Method of Wavelet Image Enhancement Based on Soft Threshold.Computer Engineering & Applications,2002,38(23):16-19.
ThePreprocessingMethodofMetabolomicMassSpectrumDataBasedontheTwo-dimensionalMaximalOverlapDiscreteWaveletTransform
Deng Kui,Li Zhenzi,Hou Yan,et al
(DepartmentofMedicalStatistics,HarbinMedicalUniversity(150081),Harbin)
ObjectiveTo preprocess metabolomic mass spectrum data through using the two-dimensional maximal overlap discrete wavelet transform(MODWT)with the purpose of removing noise and batch effects to some extent and improving the effectiveness and stability of the analytical methods.MethodsWe conducted the two-dimensional MODWT with the wavelet function of Haar to metabolomic mass spectrum data of ovarian cancer and ovarian cyst and obtained the data of different scales.And then we set the detail data to zero and reconstructed the data.After that,the random forest method was applied to the preprocessed data to screen variables,establish the discrimination model and evaluate the effects of preprocessing.ResultsThe classification performance of mass spectrum data using the two-dimensional MODWT is obviously better than the original data.ConclusionFor the mass spectrum data,the two-dimensional MODWT can well conduct feature extraction and improve the discriminant ability of the model and it has the research value and application value.
Metabolomics;Mass spectrometric data;Data preprocessing;Wavelet transform
国家自然科学基金(81302511,81473072);哈尔滨医科大学创新科学研究基金(2016JCZX13);哈尔滨医科大学卫生统计学教研室(150081)
△通信作者:李康,E-mail:likang@ems.hrbmu.edu.cn
郭海强)
