补肾蠲毒丸质量标准研究△
2017-12-26盛晓静周鹤赖杰仁
盛晓静,周鹤,赖杰仁
(1.九江市食品药品检验所,江西 九江 332000;2.九江市中医医院,江西 九江 332000)
·中药工业·
补肾蠲毒丸质量标准研究△
盛晓静1*,周鹤1,赖杰仁2
(1.九江市食品药品检验所,江西 九江 332000;2.九江市中医医院,江西 九江 332000)
目的建立补肾蠲毒丸的质量控制标准。方法采用显微鉴别法对山药、苦参、僵蚕、郁金进行定性鉴别,采用薄层色谱法对菟丝子、虎杖、黄芩、赤芍进行定性鉴别;采用高效液相色谱-蒸发光散射检测法(HPLC-ELSD)测定黄芪甲苷的含量。结果本品显微鉴别特征明显,薄层色谱定性鉴别斑点清晰、专属性强;含量测定方法中,黄芪甲苷的量在0.252 0~3.150 0 μg呈良好线性关系,相关系数r=0.999 6,平均回收率为99.80%。结论所建立的方法准确可靠、易于操作,可用于补肾蠲毒丸的质量控制。
补肾蠲毒丸;质量标准;显微鉴别;薄层色谱法;高效液相色谱-蒸发光散射检测法
补肾蠲毒丸是在九江市中医医院肝病科邹必英副主任医师的经验方基础上制成的中药浓缩丸。
制剂处方以菟丝子、枸杞子、巴戟天、肉苁蓉为君,补肾益精健脾;以虎杖、黄芩、胡黄连、青蒿、苦参为臣,清热化湿蠲毒;配以黄芪、山药、生麦芽益气健脾助运,郁金、皂角刺解郁行气化瘀,赤芍、僵蚕凉血止痛散结。全方16味补泻同用,温凉并施,益阳扶正不碍邪、达蠲邪毒不伤正,具补肾健脾、清化湿热的功能,临床用于脾肾阳虚兼夹湿热型慢性乙肝、乙肝病毒携带者的治疗多年,效果显著。制成的补肾蠲毒丸除山药、郁金、苦参、僵蚕以原粉入药外,其余12味均经过提取浓缩,具有药效缓和、作用持久、服用方便、利于携带等优点。为了有效控制制剂质量,本研究建立了山药、苦参、僵蚕、郁金、菟丝子、虎杖、黄芩、赤芍的定性鉴别及黄芪甲苷的含量测定方法。
1 仪器与试药
1.1 仪器
OLYMPUS CX41三目显微镜;Waters高效液相色谱仪(e2695分离模块、2489检测器、empower工作站);TU-1901型双光束紫外可见光分光光度计(北京普析通用仪器有限责任公司);Sartorius Bp211D 分析天平;YIY-uL 300W -C型超声波清洗机机(上海艺源超声设备有限公司);CAMAG TLC VISUALIZER 薄层色谱成像系统。
1.2 试药
超纯水、色谱乙腈(Honeywell,HPLC Grade);薄层层析用硅胶G(青岛海洋化工厂生产,化学纯);其他试剂均为分析纯;菟丝子对照药材、大黄素对照品、黄芩苷对照品、芍药苷对照品、黄芪甲苷对照品(中国食品药品检定研究院,批号分别为121232-201403、110756-200110、110715-201318、110736-201438、110781-201314,其中黄芪甲苷对照品含量以95.8%计);补肾蠲毒丸(九江市中医医院制剂室,批号分别为20141001、20141002、20141003、20150101、20150102、20150103、20150104、20150701、20150702、20150703)。
2 方法与结果
2.1 显微鉴别
本品粉末棕褐色。草酸钙针晶束存在于黏液细胞中,针晶长80~240 μm,粗2~5 μm(山药);纤维束周围的细胞中含草酸钙方晶,形成晶纤维,含晶细胞的壁不均匀木化增厚(苦参);菌丝体存在于体壁或淡棕色、半透明结晶块中,菌丝近无色,细长,直径1~5 μm,相互盘缠交织(僵蚕);含糊化淀粉粒的薄壁细胞单个散在或数个相连(郁金)[1-2]。见图1。
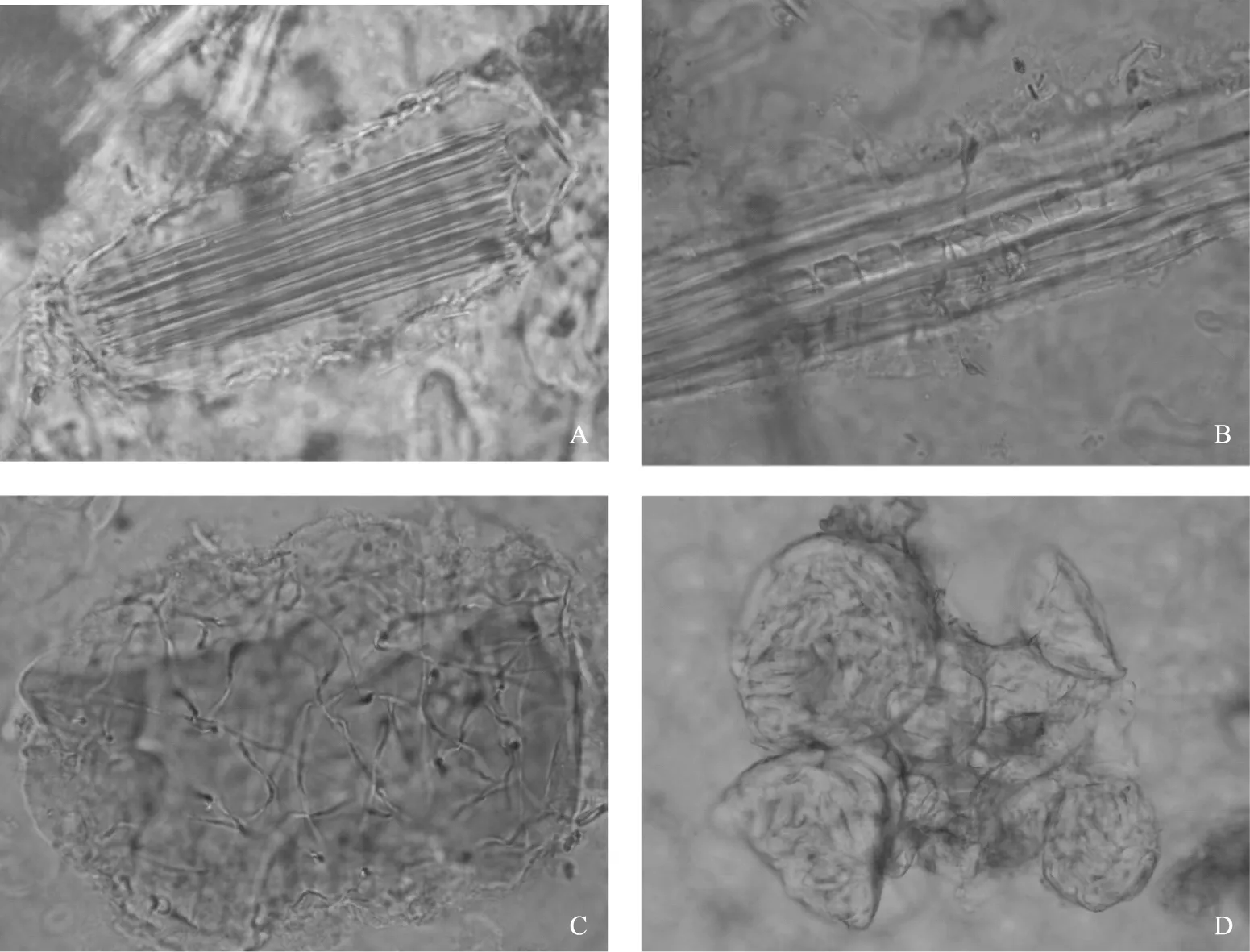
注:A.草酸钙针晶束(山药);B.晶纤维(苦参);C.菌丝体(僵蚕);D.含糊化淀粉粒的薄壁细胞(郁金)。图1 补肾蠲毒丸粉末显微特征图(10×40倍)
2.2 薄层色谱鉴别
2.2.1 菟丝子的鉴别 取补肾蠲毒丸样品2 g,研细,加水30 mL,搅匀,用脱脂棉滤过,滤液置分液漏斗中,加石油醚(30~60 ℃)30 mL,振摇提取,弃去石油醚提取液,加乙酸乙酯振摇提取2次,每次30 mL,合并乙酸乙酯提取液,蒸干,残渣加甲醇2 mL使溶解,作为供试品溶液。另取菟丝子对照药材0.5 g,加水30 mL,煎煮30 min,放冷,滤过,滤液置分液漏斗中,同法制成对照药材溶液。再取缺菟丝子阴性对照样品2 g,与供试品溶液的制备方法同法制得阴性对照样品溶液。吸取上述3种溶液各5 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲酸-水(10∶1∶2)的上层溶液为展开剂,展开,取出,晾干,置紫外灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,阴性对照样品无干扰[3]。见图2。
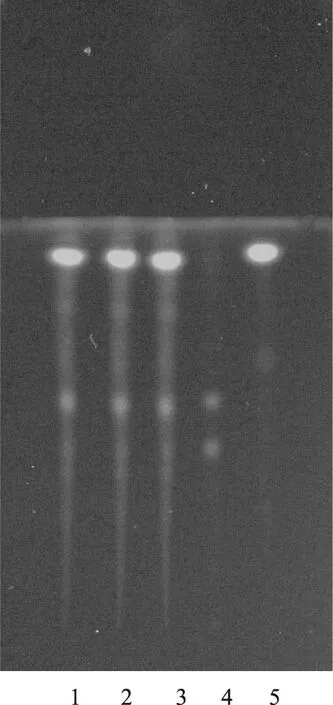
注:1~3.补肾蠲毒丸样品;4.菟丝子对照药材;5.缺菟丝子的阴性对照。图2 补肾蠲毒丸中菟丝子TLC鉴别图

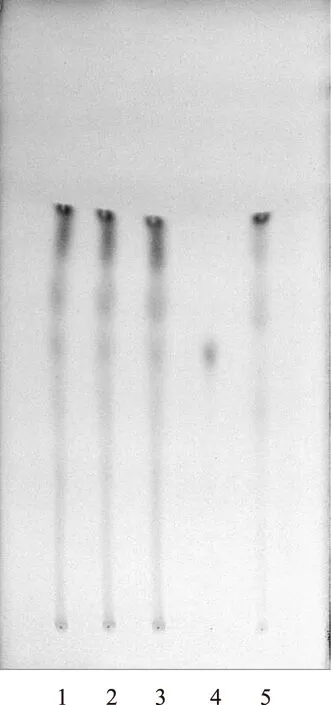
注:1~3.补肾蠲毒丸样品;4.大黄素对照品;5.缺虎杖的阴性对照。图3 补肾蠲毒丸中虎杖TLC鉴别图
2.2.2 虎杖的鉴别 取补肾蠲毒丸样品5 g,研细,加甲醇30 mL,超声处理30 min,滤过,滤液蒸干,残渣加水20 mL使溶解,再加盐酸2 mL,水浴加热30 min,放冷,用乙醚振摇提取3次,每次15 mL,合并乙醚液,蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取大黄素对照品,加甲醇制成质量浓度为1 mg·mL-1的溶液,作为对照品溶液。再取缺虎杖的阴性对照样品5 g,与供试品溶液的制备方法同法制得阴性对照样品溶液。吸取上述供试品溶液及阴性对照样品溶液各5 μL、对照品溶液2 μL,分别点于同一硅胶G薄层板上,以石油醚(30~60 ℃)-甲酸乙酯-甲酸(15∶5∶1)的上层溶液为展开剂,展开,取出,晾干,置氨气中熏至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,阴性对照样品无干扰[4]。见图3。
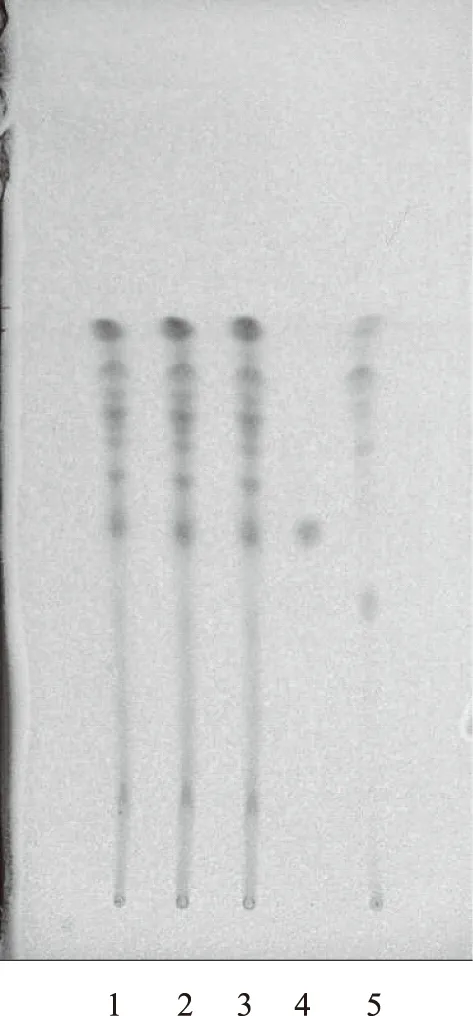
2.2.3 黄芩的鉴别 取补肾蠲毒丸样品5 g,研细,加乙酸乙酯-甲醇(3∶1)的混合溶液30 mL,加热回流30 min,放冷,滤过,滤液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取黄芩苷对照品,加甲醇制成质量浓度为1 mg·mL-1的溶液,作为对照品溶液。再取缺黄芩的阴性对照样品5 g,与供试品溶液的制备方法同法制得阴性对照样品溶液。吸取上述3种溶液各5 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-丁酮-甲酸-水(5∶3∶1∶1)为展开剂,展开,取出,晾干,喷以5%三氯化铁乙醇溶液。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,阴性对照样品无干扰[5]。见图4。
注:1~3.补肾蠲毒丸样品;4.黄芩苷对照品;5.缺黄芩的阴性对照。图4 补肾蠲毒丸中黄芩TLC鉴别图
2.2.4 赤芍的鉴别 取补肾蠲毒丸样品5 g,研细,加甲醇30 mL,超声处理30 min,滤过,滤液蒸干,残渣加水20 mL使溶解,用水饱和的正丁醇振摇提取3次,每次20 mL,合并正丁醇液,用正丁醇饱和的水洗涤3次,每次20 mL,取正丁醇液蒸干,残渣加甲醇2 mL使溶解,作为供试品溶液。另取芍药苷对照品,加甲醇制成质量浓度为1 mg·mL-1的溶液,作为对照品溶液。再取缺赤芍的阴性对照样品5 g,与供试品溶液的制备方法同法制得阴性对照样品溶液。吸取上述3种溶液各5 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13∶7∶2)的下层溶液为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,在105 ℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,阴性对照样品无干扰[6-7]。见图5。
注:1~3.补肾蠲毒丸样品;4.芍药;苷对照品;5.缺赤芍的阴性对照。图5 补肾蠲毒丸中赤芍TLC鉴别图
2.3 含量测定
2.3.1 色谱条件与系统适用性试验 色谱柱:Thermo SCIENTIFIC HypersiL GOLD(250 mm×4.6 mm,5 μm);柱温:25 ℃;流动相为乙腈-水(31∶69V/V);流速:1.0 mL·min-1;蒸发光散射检测器:雾化器温度35 ℃;漂移管温度80 ℃;载气N2;气体压力0.207 MPa。理论塔板数按黄芪甲苷峰计算应不低于7000。
2.3.2 对照品溶液的制备 取黄芪甲苷对照品适量,精密称定,加甲醇制成质量浓度为0.13 mg·mL-1的溶液,即得。
2.3.3 供试品溶液的制备 取本品适量,研细,取约10 g,精密称定,置索氏提取器中,加甲醇100 mL,加热回流提取6 h,提取液回收溶剂并浓缩至干,残渣加水25 mL,微热使溶解,用水饱和的正丁醇振摇提取4次,每次25 mL,合并正丁醇提取液,用浓氨试液洗涤2次,每次25 mL,浓氨试液再用水饱和的正丁醇25 mL提取,合并正丁醇提取液,回收正丁醇至干,残渣用甲醇溶解并转移至10 mL量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得[8]。
2.3.4 测定方法 分别精密吸取对照品溶液10、20 μL,供试品溶液10 μL,注入液相色谱仪,测定,用外标两点法对数方程计算,即得。
2.3.5 阴性对照试验 取按照本品制剂处方及生产工艺要求制备的缺黄芪的阴性样品,按2.3.3项下供试品溶液的制备方法处理,即得缺黄芪的阴性对照样品溶液,分别精密吸取对照品溶液、供试品溶液及阴性对照样品溶液各10 μL进行高效液相色谱试验,所得阴性对照色谱图中,在补肾蠲毒丸色谱图中黄芪甲苷的出峰位置上未出现色谱峰,表明处方中其他药材对黄芪甲苷的含量测定无干扰。见图6。
2.3.6 线性关系的考察 精密吸取黄芪甲苷对照品溶液(0.126 0 mg·mL-1)2、5、10、15、20、25 μL,分别注入液相色谱仪,测定其峰面积,以黄芪甲苷进样量的常用对数为横坐标,以峰面积的常用对数为纵坐标,绘制标准曲线,计算回归方程:Y=1.633X+6.516,r=0.999 6。结果表明,黄芪甲苷的进样量在0.252 0~3.150 0 μg呈良好的线性关系。
2.3.7 精密度试验 精密吸取同一对照品溶液10 μL,重复进样6次,按2.3.1项下色谱条件,测定峰面积分别为4 658 951、4 691 602、4 480 464、4 800 282、4 580 005、4 629 281,对应的常用对数值分别为6.668、6.671、6.651、6.681、6.661、6.666,RSD为0.16%,结果表明仪器的精密度良好。
2.3.8 稳定性试验 精密吸取供试品溶液10 μL,分别于0、2、4、6、8、24 h,注入高效液相色谱仪,测定黄芪甲苷含量,RSD为0.60%,结果表明供试品溶液在24 h内稳定。
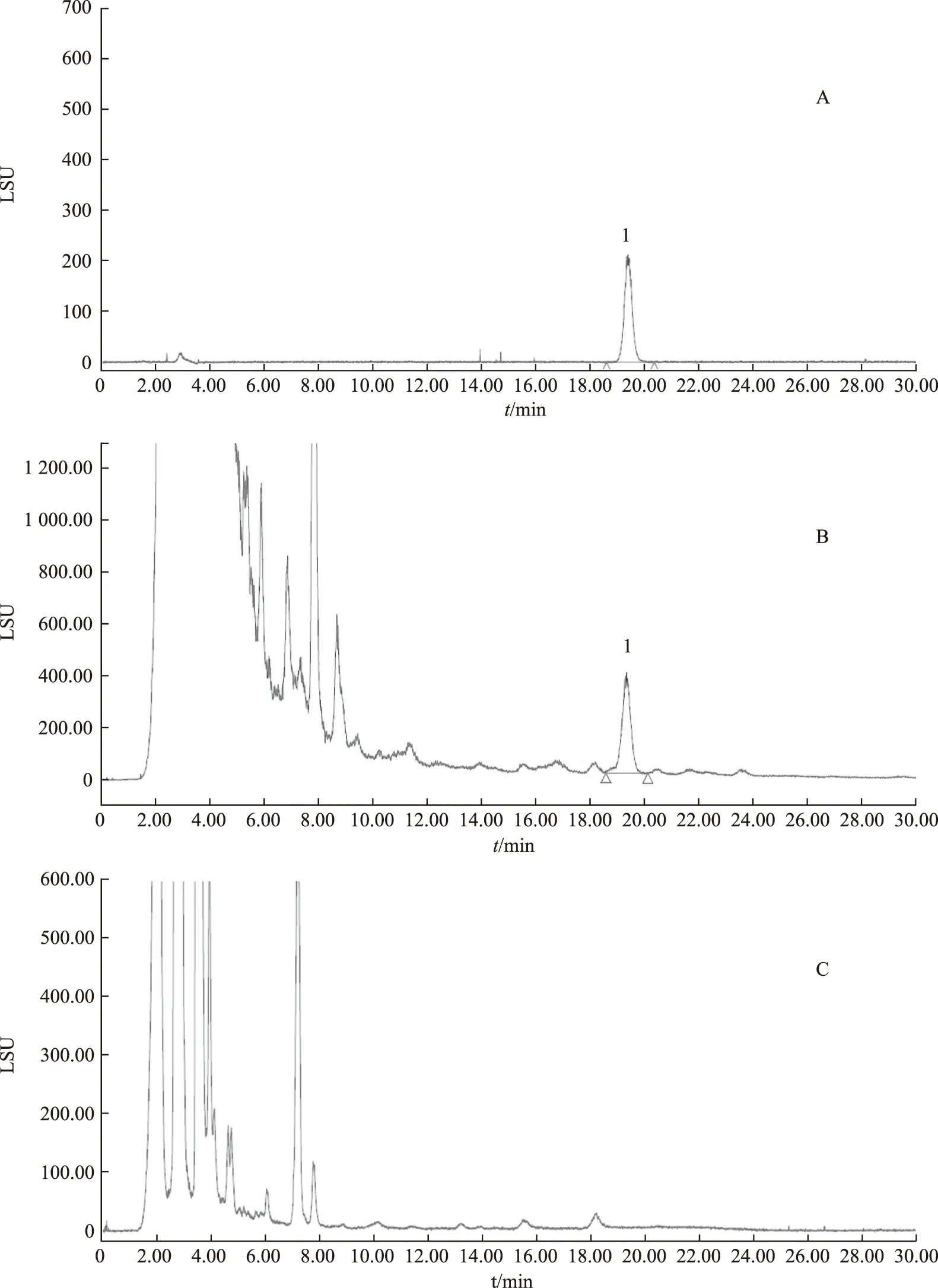
注:A.黄芪甲苷对照品;B.样品;C.阴性对照品;1.黄芪甲苷。图6 黄芪甲苷对照品、样品及阴性对照品HPLC图
2.3.9 重复性试验 取同一批号样品(20150101)6份,按2.3.3项下方法平行操作后,分别测定黄芪甲苷含量,结果含量平均值为0.194 5 mg·g-1,RSD为1.39%。结果表明,在同一条件下,6次测得样品黄芪甲苷含量一致。
2.3.10 回收率试验 采用加样回收法,精密称取已知含量的补肾蠲毒丸样品(20150101)6份,分别精密添加一定量的黄芪甲苷对照品,按2.3.3项下方法制成供试品溶液,各取10 μL进样,测定黄芪甲苷含量,结果见表1。
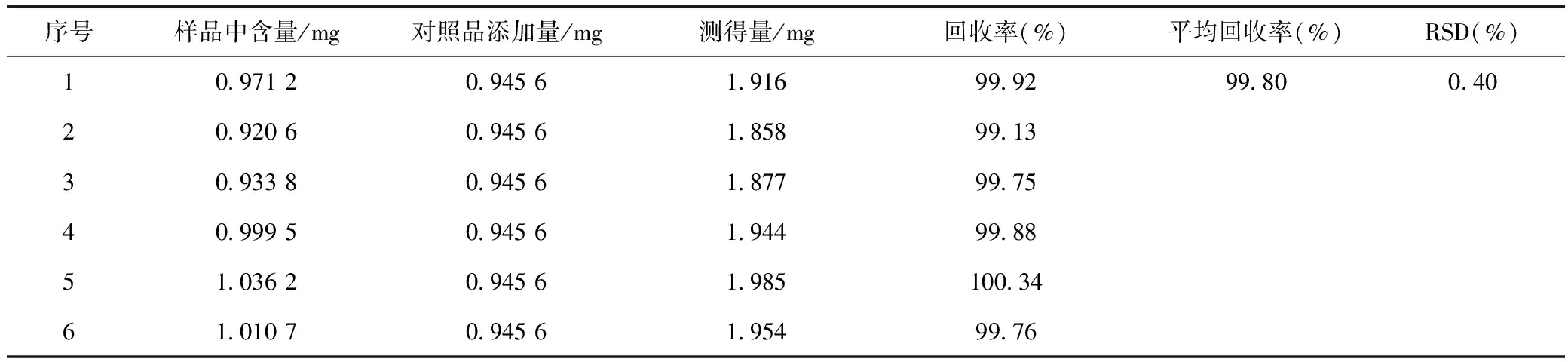
表1 补肾蠲毒丸中黄芪甲苷回收率测定(n=6)
2.3.11 样品测定 取10批样品,按上述色谱条件和方法测定黄芪甲苷的含量,测定结果见表2。
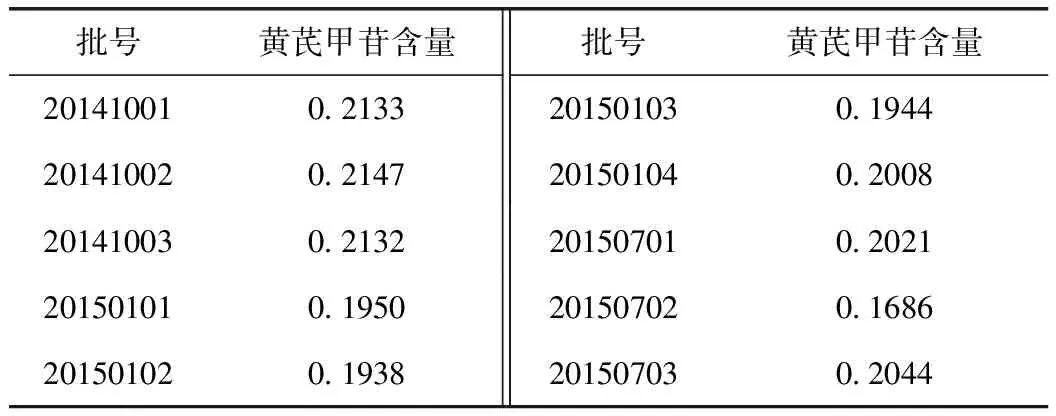
表2 10批补肾蠲毒丸含量测定结果 mg·g-1
3 讨论
补肾蠲毒丸处方配伍16味,其中山药、苦参、僵蚕、郁金4味以药材原粉入药,采用显微鉴别法简单易行,可有效定性鉴别。通过薄层色谱法实验研究,菟丝子、虎杖、黄芩、赤芍均可采用适宜的提取方法和展开系统进行定性鉴别,且薄层色谱斑点明显,阴性对照无干扰。
考虑到本品重用黄芪补气、利水,用量占处方总药量的14.6%,我们研究了采用高效液相色谱法测定黄芪甲苷的含量。取黄芪甲苷对照品溶液进行紫外光谱扫描,结果最大吸收峰在200 nm波长处属于末端吸收,故选择用蒸发光散射检测器(ELSD)进行检测。选择不同品牌的十八烷基硅烷键合硅胶柱进行实验,结果当流动相为乙腈-水(31∶69)时,采用Waters SunFireTMC18、Thermo HypersiL GoLd或SHIMADZU-GL WondaCr act ODS-2等色谱柱时,均能达到有效分离的目的,且峰形较好,阴性对照样品无干扰。供试品溶液的制备主要包括索氏提取和萃取两部分,为了优化供试品溶液的制备方法,分别考察了索氏提取4、6、8 h后萃取4次以及索氏提取6 h后萃取3、4、5次的含量测定值,结果显示索氏提取6 h后萃取4次已经能够充分提取待测成分。
在实验过程中,还参考文献方法[9-10]对处方中黄芪、枸杞子进行了薄层色谱研究,结果在与对照药材色谱相应的位置上,缺黄芪的阴性对照样品除黄芪甲苷外,其他主斑点均有干扰,缺枸杞子的阴性对照样品主斑点有干扰,未能进行定性鉴别。
[1] 徐国均,徐珞珊,刘柏英,等.中药材粉末显微鉴定[M].北京:人民卫生出版社,1986.
[2] 陈俊华,舒光明,姜荣兰.中药粉末显微鉴别手册:第一卷[M].重庆:四川省中药研究所,1985.
[3] 彭维,郑文燕,苏畅,等.安神补心颗粒质量标准提高研究究[J].中药材,2013,36(8):1343-1347.
[4] 国家药典委员会.中华人民共和国药典:一部[S].北京:中国医药科技出版社,2010.
[5] 傅应华,徐宏祥.消瘰合剂的质量控制项目研究[J].药物分析杂志,2010,30(3):526-529.
[6] 于红艳,许成刚,崔永霞.退黄丸的质量标准研究[J].中成药,2013,35(1):79-86.
[7] 程东岩,王隶书,程东红,等.赤丹肝纤颗粒的质量标准[J].中国药师,2015,18(6):1027-1030.
[8] 聂颖兰,范斌,郭娜,等.HPLC-ELSD 法测定健脾益肾胶囊中黄芪甲苷的含量[J].中国实验方剂学杂志,2013,19(17):130-132.
[9] 张凤瑞,周贤,刘炎,等.当归补血汤中黄芪的鉴别及黄芪甲苷的含量测定[J].吉林中医药,2013,33(3):289-291.
[10] 唐柏平,李琼英,贺清源.阴阳和口服液的薄层色谱实验研究[J].湖南中医杂志,2012,28(6):116-118.
StudyonQualityStandardofBushenJuanduPills
SHENG Xiaojing1*,ZHOU He1,LAI Jieren2
(1.JiujiangInstituteforFoodandDrugControl,Jiujiang332000,Jiangxi,China; 2.JiujiangCityHospitalofTraditionalChineseMedicine,Jiujiang332000,Jiangxi,China)
Objective:To establish the quality standard for Bushen Juandu Pills.MethodsThe Dioscoreae Rhizoma,Sophorae Flavescentis Radix,Bombyx Batryticatus and Curcumae Radix were identified by microscopic identification.The Cuscutae Semen,Polygoni Cuspidati Rhizoma et Rradix,Scutellariae Radix and Paeoniae Radix Rubra were identified by TLC,and the content of astragaloside was determined by HPLC-ELSD.ResultsThe microscopic characteristics were obvious,TLC spots were clear and strong specific.In the content determination method,astragaloside showed a good linear relationship at the range of 0.252 0-3.150 0 μg,r=0.999 6.The average recovery rate was 99.80%.ConclusionThe established method is accurate and reliable and easy to operate,it can be used for the quality control of Bushen Juandu pills.
Bushen Juandu Pills;quality standard;microscopic identification;TLC;HPLC-ELSD
10.13313/j.issn.1673-4890.2017.9.019
九江市社会发展与软科学类科研计划项目(九科字[2015]44号-20)
*
盛晓静,副主任中药师,研究方向:中药质量分析;TeL:(0792)8157328,E-maiL:shengxj11@163.com
2016-10-25)
