人杀菌/渗透增强蛋白N端功能片段基因突变文库的构建和鉴定
2017-12-19李晶琴孔庆利安云庆
李晶琴 孔庆利 安云庆*
(1.首都医科大学燕京医学院医学检验学系,北京 101300;2.首都医科大学基础医学院免疫学系,北京 100069)
·基础研究·
人杀菌/渗透增强蛋白N端功能片段基因突变文库的构建和鉴定
李晶琴1孔庆利2安云庆2*
(1.首都医科大学燕京医学院医学检验学系,北京 101300;2.首都医科大学基础医学院免疫学系,北京 100069)
目的为提高杀菌/渗透性增强蛋白N端功能片段BPI23对内毒素(lipopolysaccharides,LPS)的亲和性,通过易错聚合酶链式反应(polymerase chain reaction,PCR)和DNA改组技术,对其编码序列BPI600进行突变,在大肠杆菌XL10-Gold中构建BPI600基因突变体库。方法通过控制Mg2+和Mn2+的浓度进行易错PCR,获得BPI600随机突变基因片段,经测序和基因比对,确定BPI600的随机突变率。再对易错PCR产物进行DNA改组,将改组产物克隆至pYD1载体中,转化大肠杆菌XL10-Gold,从Amp抗性(Luria-Bertani,LB)固体培养平板上任选10个单菌落,增菌后提取质粒,得到pYD1-shuffledBPI600重组质粒,将质粒进行HindⅢ/XhoⅠ酶切鉴定,取5个阳性质粒进行DNA测序,通过基因和氨基酸比对鉴定突变情况。结果测序和基因比对显示,BPI600经易错PCR所获突变文库的随机突变率达2.3%;改组后重组质粒的酶切结果表明,在随机选择的10个菌落所提质粒中,有6个含BPI600,表明构建了重组质粒pYD1-shuffledBPI600;在5个阳性质粒中,有4个重组质粒的BPI23编码序列分别发生了6、9、11和14个碱基突变;1个不仅有10个碱基突变,还存在1个碱基的缺失。氨基酸比对表明,有2个质粒(分别有6和14个碱基突变)具有正确的读码框,能够翻译完整的蛋白质,各有4或14个氨基酸突变。3个质粒因含终止密码子而不能表达完整目的蛋白。结论成功构建pYD1-shuffledBPI600重组质粒,获得库容量为2×105的突变文库,为高内毒素亲和力突变体的筛选奠定了基础。
人杀菌/渗透增强蛋白;定向进化;易错PCR;DNA改组
杀菌/渗透增强蛋白(bactericidal/permeability increasing protein,BPI)是生物体内存在的一种阳离子抗菌蛋白,BPI及其功能性N端片段BPI23具有广谱杀伤包括耐药菌株在内的革兰阴性菌以及中和内毒素的作用[1],在临床上具有较大的应用前景。
为进一步提高BPI23与内毒素的亲和性和抗感染效果,本实验在前期构建的pYD1-BPI600重组质粒的基础上,对BPI N端功能片段BPI23的编码序列BPI600进行定向进化。蛋白质定向进化可以模拟自然进化的过程(随机突变或重组),通过人为地改变反应条件,在体外进行基因的突变或重组,以构建突变体库并从中筛选出理想突变体。易错PCR(error-prone polymerase chain reaction,EP-PCR)和DNA改组(DNA shuffling)是目前常用的定向进化技术[2]。易错PCR基于Taq DNA聚合酶不具备3′→5′外切酶活性,缺乏校对功能,故在DNA扩增过程中会发生错误掺入而引起突变[3]。DNA改组是将亲本基因进行尽可能多的组合,最终获得大容量基因突变体库的技术[4]。本实验通过易错PCR和DNA改组技术,构建了大容量的BPI600突变体库,为优势突变体的筛选打下基础。
1 材料与方法
1.1 材料
1.1.1 质粒和菌株
大肠杆菌DH5α由本室保存,大肠杆菌XL10-Gold超级感受态细胞购自美国Stratagene公司,pMD18-T载体购自日本TaKaRa公司,质粒pYD1-BPI600由本室构建,包含BPI600基因(约600 bp)。载体pYD1全长5 009 bp,购自美国Invitrogen公司。
1.1.2 主要试剂
dCTP、dTTP、dATP、dGTP、Taq DNA 聚合酶、MnCl2、MgCl2购自上海生工生物工程公司。NZ胺购自美国Amresco公司、β-巯基乙醇(β-ME)购自美国Stratagene公司。限制性内切酶BamHI、HindⅢ购自日本TaKaRa公司。100 bp Ladder Marker、DL2000 Marker购自中国BioDev公司。DL15000购自日本TaKaRa公司。DNA小片段胶回收试剂盒购自中国BioDev公司。质粒大量提取试剂盒购自威格拉斯生物技术有限责任公司。其余试剂为国产分析纯。
1.1.3 引物设计与合成
参照GenBank中BPI基因的序列,设计扩增BPI600的引物,P1:5′-CCC AAG CTT GTC AAC CCT GGC GTC GTG-3′,添加HindⅢ酶切位点(下划线标示),P2:5′-CCG CTC GAG TCA TAT TTT GGT CAT TAC TGG-3′,添加XhoⅠ酶切位点(下划线标示);P3:5′-AGTAACGTTTGTCAGTAATTGC-3′,P4:5′-GTCGA TTTTGTTACATCT ACAC-3′,为载体pYD1测序引物;P5:5′-GTC AAC CCT GGC GTC GTG-3′,P6:5′-TAT TTT GGT CAT TAC TGG-3′为易错PCR设计引物。P1/P2、P5/P6由上海生工生物工程公司合成;P3/P4购自美国Invitrogen公司。
1.2 易错PCR
1.2.1 随机突变片段的获得
在高Mg2+离子浓度(5.0 mmol/L)和Mn2+存在的条件下,通过选择不同的Mg2+浓度来控制随机突变的频率。以质粒pYD1-BPI600为模板进行易错PCR。反应体系为(50 μL):10×EP-PCR 缓冲液5 μL,dNTP混合物(各10 mmol/L)1 μL,P5、P6引物(均为10 μmol/L)各2 μL,质粒模板1 μL,MnCl2(5 mmol/L)5 μL,Taq DNA 聚合酶(5 u/μL)0.25 μL,去离子水加至50 μL。扩增条件为:95℃预变性5 min;94℃ 1 min、55℃ 1 min、72 ℃ 1 min,共30个循环。用1%(质量分数)琼脂糖凝胶电泳分析扩增产物,并在紫外灯下分离出含大小约600 bp基因片段的凝胶,利用DNA小片段胶回收试剂盒纯化回收BPI600随机突变片段;以回收物为模板,再行第二轮易错PCR,按上述方法回收纯化大小约为600 bp突变基因片段。
1.2.2 突变基因的克隆和鉴定
将纯化的易错PCR产物连接至克隆载体pMD18-T中,转化大肠杆菌DH5α感受态细胞。利用Amp抗性(Luria-Bertani,LB)固体培养基进行筛选,菌落PCR鉴定是否含插入片段,并从含插入片段的阳性菌落中随机挑取5个菌落进行测序和基因比对,并计算随机突变率。
1.3 DNA改组
1.3.1 随机突变基因片段化处理
将易错PCR获得的随机突变基因片段利用超声波进行碎片化处理,条件为:功率设为4 W,冰浴下超声6 s,间隔12 s,循环处理400次。产物经2%(质量分数)琼脂糖凝胶电泳,切下大小为50~100 bp左右的条带,用DNA小片段回收试剂盒进行回收纯化。
1.3.2 无引物PCR
反应体系(20 μL)为:10×PCR buffer 2 μL,dNTP(10 mmol/L)0.5 μL,MgCl2(25 mmol/L)1.2 μL,DNA小片段10 μL,Taq DNA 聚合酶(5 u/μL)0.25 μL,加超纯水至20 μL。反应条件为:95℃预变性5 min;94 ℃ 30 s、45 ℃ 30 s、72 ℃ 30 s,共50个循环;72 ℃延伸10 min。用1%(质量分数)琼脂糖凝胶电泳鉴定反应产物片段大小。
1.3.3 有引物PCR
将上述无引物PCR产物1∶20稀释后,作为模板进行有引物PCR扩增。反应体系(20 μL)为:无引物PCR产物稀释液5 μL,10×PCR buffer 2 μL,dNTP(10 mmol/L)1 μL,MgCl2(25 mmol/L)3 μL,P1和P2各1 μL,Taq DNA 聚合酶(5 μ/μL)0.25 μL,加超纯水至50 μL。扩增条件为:95℃预变性5 min;94 ℃ 60 s、57 ℃ 60 s、72 ℃ 75 s,共35个循环;72℃延伸5 min。扩增产物经1%(质量分数)琼脂糖凝胶电泳分离后,在紫外灯下切下约600 bp的凝胶条带,用DNA小片段胶回收试剂盒回收、纯化BPI600的改组片段。
1.4 重组质粒文库的构建和鉴定
1.4.1 重组质粒文库的构建
BPI600改组基因片段和质粒载体pYD1分别用HindⅢ/XhoⅠ酶切,产物分别经1%(质量分数)琼脂糖凝胶电泳分离后,在紫外灯下切下含目的基因片段的凝胶,用DNA小片段胶回收试剂盒回收纯化酶切产物并进行连接,反应体系为:质粒载体pYD1的酶切产物0.5 μL,BPI600改组基因的酶切产物7.5 μL,10×连接缓冲液1 μL,T4 DNA连接酶1 μL,总体积10 μL;16 ℃孵育12 h。用预冷枪头取大肠杆菌XL10-Gold超级感受态细胞100 μL、β-ME 4 μL放入预冷的Falcon聚丙烯离心管中,轻轻混匀后冰浴10 min;再加入1 μL上述连接产物,轻轻旋转使感受态细胞与连接产物混匀,冰浴30 min;将聚丙烯离心管置于 42 ℃水浴中作用30 s后,立即放入冰浴中作用2 min;再加入预热的NZY+肉汤900 μL,37 ℃、225 r/min培养1 h后,取100 μL菌液涂布于Amp抗性LB固体培养基上;37 ℃培养12~16 h,获得Amp抗性阳性克隆菌,计数菌落数。
1.4.2 重组质粒文库的鉴定
从Amp抗性LB固体培养基上随机挑取10个单菌落,增菌、提取质粒后进行酶切鉴定;再从经鉴定含有插入片段的阳性转化菌落所提质粒中随机选取5个,在pYD1测序引物P3和P4的引导下进行测序分析。测序结果与GenBank中的野生型BPI600序列进行DNA比对,分析pYD1-shuffledBPI600重组质粒中目的基因的突变情况。
2 结果
2.1 易错PCR结果
1%(质量分数)琼脂糖凝胶电泳证实两轮易错PCR均成功扩增出大小约为600 bp的基因片段(图1)。
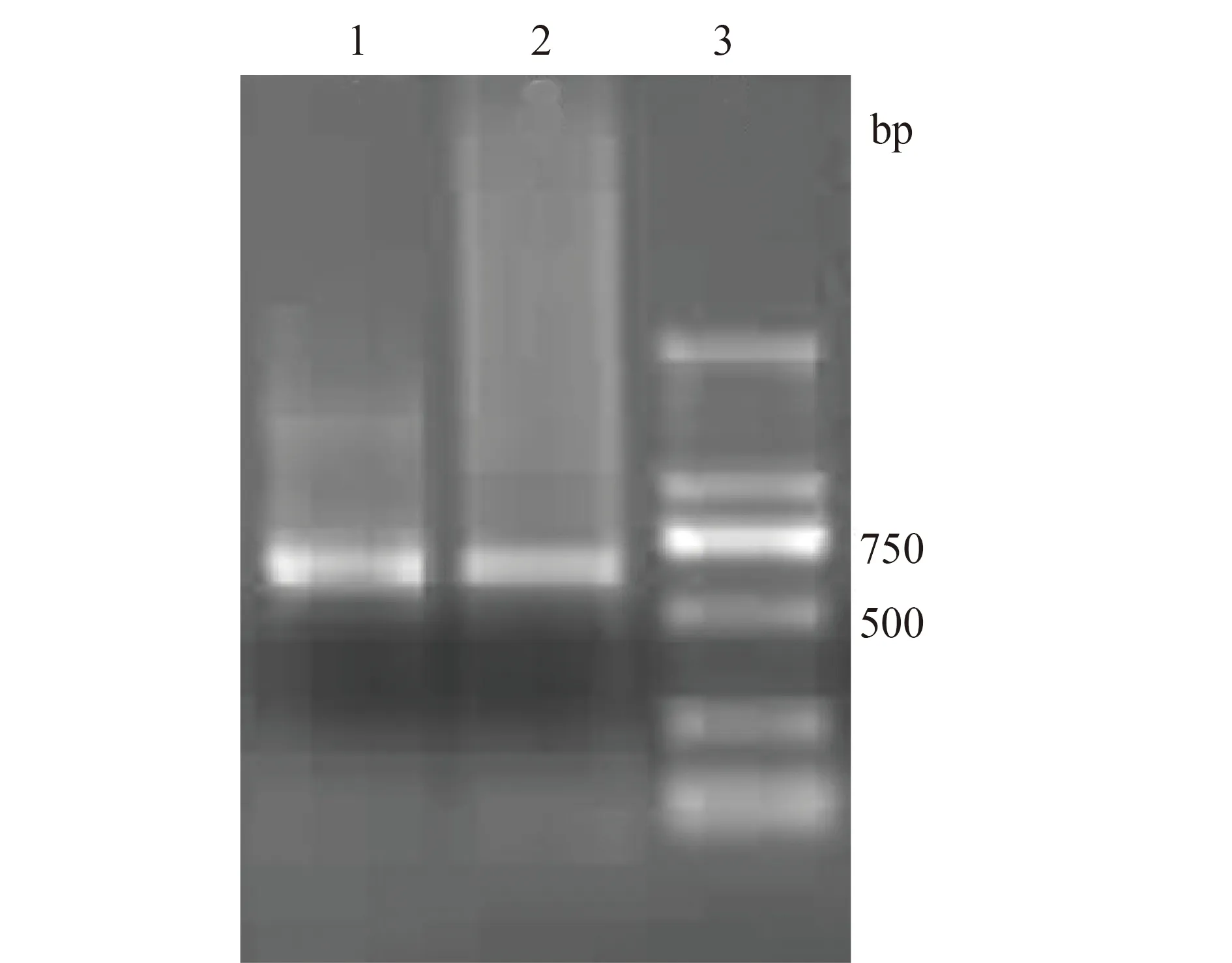
图1 易错PCR扩增产物Fig.1 The product of EP-PCR
1:product of the first EP-PCR;2:product of the second EP-PCR;3:DL 2 000 marker;EP: error prone;PCR: polymerase chain reaction
从Amp抗性LB固体培养基筛选出的BPI600的随机突变基因库中,随机挑选经菌落PCR鉴定为阳性的5个菌落进行测序,测序结果与野生型BPI600基因进行比对(图2),经过计算,BPI600随机突变库的随机突变率达2.3%(68/3 000)。
2.2 DNA改组结果
对易错PCR产物进行碎片化处理后,获得50~100 bp左右的随机片段,经50个循环的无引物PCR,小片段DNA被拼接成许多大小不一的改组分子,再经过35个循环的有引物PCR,重聚到与原基因分子大小相同的BPI600改组分子(图3)。
2.3 pYD1-shuffled BPI600重组质粒文库的构建与鉴定结果
将有引物PCR产物克隆至pYD1载体中,构建重组质粒pYD1-shuffledBPI600,并用其转化大肠杆菌XL10-Gold,在Amp抗性LB固体培养平板上获得Amp抗性阳性克隆菌,计数菌落数,获得了2×105个转化子。
从上述平板上随机选取10个单菌落,增菌后提取质粒,用HindⅢ和XhoⅠ酶切,在10个质粒中,6个被酶切为大小约为4 841 bp和600 bp的两个条带,表明重组质粒中插入了BPI600片段(图4)。
将酶切鉴定后的5个阳性克隆进行测序分析,与NCBI GenBank中野生型BPI编码序列比对,其中第1、2、4、5号质粒分别发生9、14、6、11个碱基突变(图5);第3号质粒不仅有10个碱基突变,还存在一个碱基的缺失(图6)。氨基酸序列比对结果(图7)表明,在5个克隆中,第2、4号质粒具有正确的读码框,能够翻译完整的BPI23,且分别存在14和4个氨基酸突变;第1、3、5号质粒因含终止密码子而不能表达完整的目的蛋白。

图2 5个易错PCR阳性克隆的基因序列与野生型的比对结果Fig.2 Genetic comparison of 5 positive clones (after EP-PCR) with wild type
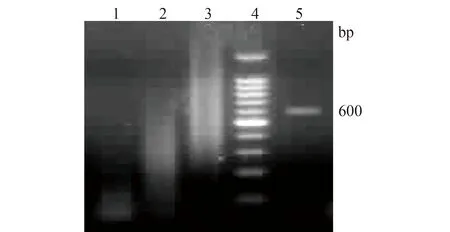
图3 BPI600 DNA改组结果Fig.3 The product of BPI600 shuffling
1:fragmented fragments;2,3:product of primerless PCR;4:100 bp Ladder marker;5:product of PCR with primers;PCR:polymerase chain reaction.
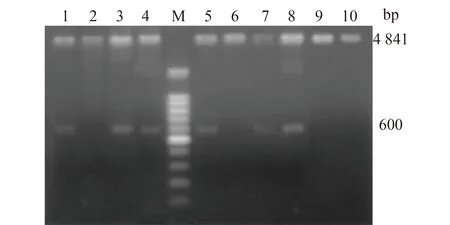
图4 pYD1-shuffled BPI600重组质粒的酶切鉴定 Fig.4 Double digestion of recombinant plasmid pYD1-shuffled BPI600 by HindⅢ/XhoⅠ
1,3,4,5,7,8:positive clones;2,6,9,10:negative clones;M:100 bp Ladder marker
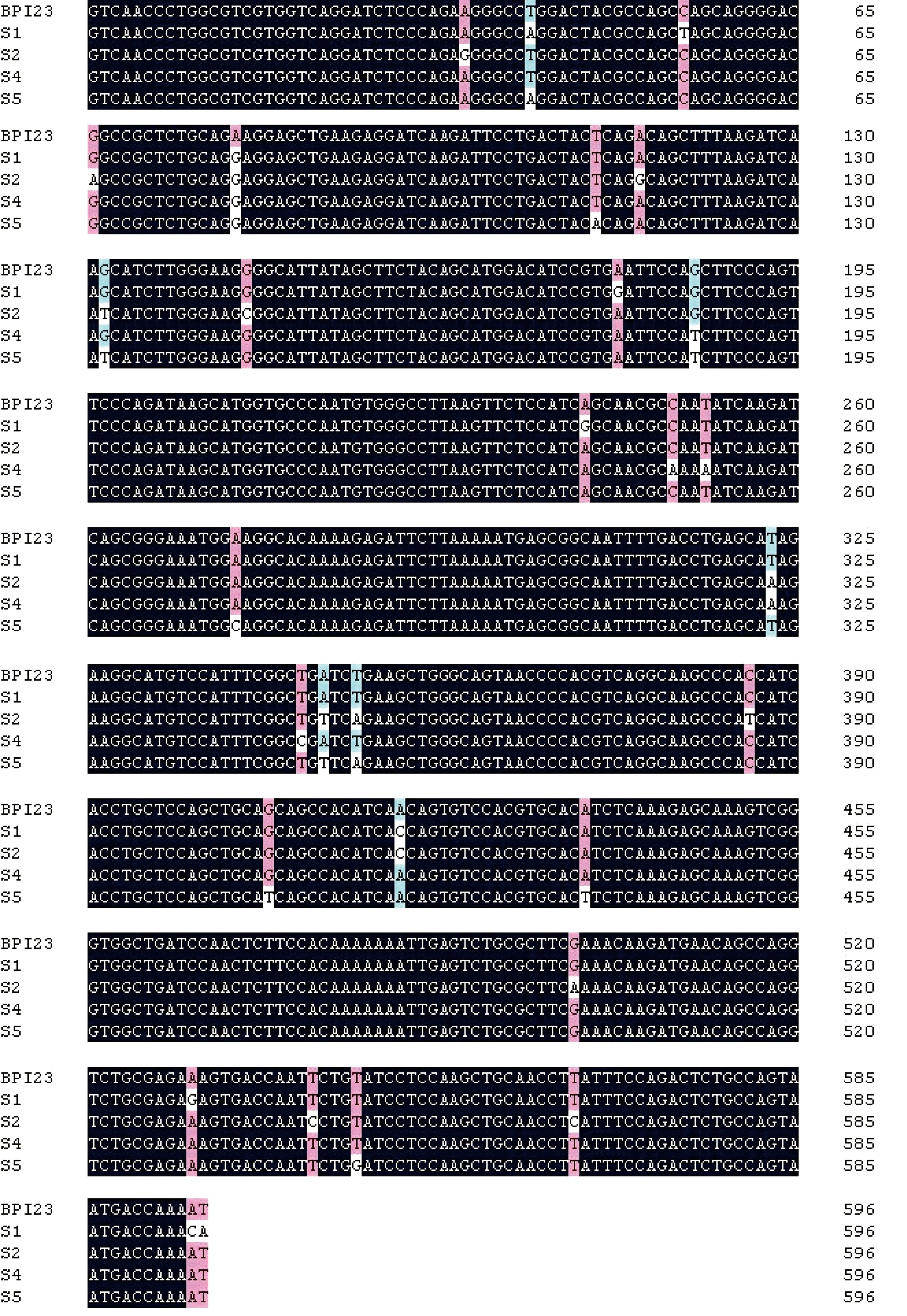
图5 1、2、4、5号克隆的基因序列与野生型的比对结果Fig.5 Genetic comparison of the number 1,2,4,5 clone with wild type

图6 3号克隆的基因序列与野生型的比对结果Fig.6 Genetic comparison of the number 3 clone with wild type
3 讨论
蛋白质的体外定向进化又称实验分子进化,因无需事先了解蛋白的空间结构和作用机制,所以几乎适用于任何蛋白质分子的改造和优选,为研究蛋白质结构与功能之间的关系提供了一种更方便、更快捷的新途径[2]。定向进化是否成功主要受以下3个方面的影响:①合理的进化策略;②合适的表达系统;③高通量的筛选方法。
在前期的实验中,研究组已成功构建了BPI23的酿酒酵母细胞表面展示系统,该系统能表达功能性BPI23,并可用流式细胞仪进行快速鉴定和高通量分选[5],适合于大容量突变体库的筛选和BPI23的进化研究。但有了合适的表达系统和高通量的筛选方法,还必须在有足够大的突变体库构建成功的基础上才能发挥作用。
易错PCR是一种简便易行的蛋白质定向进化策略,已广泛应用于蛋白质的体外进化,如酶活性的提高[6-8]、酶热稳定性的改良[9]等。在本实验中,通过控制Mg2+或Mn2+的浓度进行连续易错PCR的方法[10],成功进行了BPI600的随机突变,突变率在2.3%,较为适于后续优势株的筛选[11]。到目前为止,尚未见到BPI亚型的报道,因此选择易错PCR产生的随机突变库作为进化的出发点,可能是避免异源基因导入的一个有效的方法。DNA改组是用DNaseI消化或超声处理多条具有同源序列的基因(一般同源性>70%)或基因家族[12]。

图7 2、4号改组克隆的氨基酸序列与野生型的比对结果Fig.7 Amino acid comparison of the number 2,4 clone with wild type
产生基因随机裂解片段后,先用无引物PCR进行基因重排,然后再进行有引物PCR扩增全长基因可获得大容量基因突变体库,目前DNA改组技术广泛应用于基因突变体库的构建,大容量的突变库再结合高通量的筛选方法,便于优势突变体的筛选[13-15]。本实验在易错PCR产生随机突变的基础上进行DNA改组,与采用单纯的随机突变或定点突变方法构建的突变库相比,产生的库容量大、多样性好,可有效积累更多的有益突变,同时也可实现同源序列间的大片段交换,从而达到目的蛋白多种特性共进化的目的。实验结果表明,构建的pYD1-shuffledBPI600重组质粒中含有能够翻译为完整BPI23突变体的DNA片段,但文库中也存在一些突变克隆,因含终止密码子,最终只能翻译BPI23的部分氨基酸片段。本实验成功构建了库容量达2×105的BPI600突变体库和pYD1-shuffledBPI600重组质粒文库,为下一步筛选优势突变体的研究提供了新途径。
[1] Wilde C G,Seilhamer J J,McGrogan M,et al.Bactericidal/permeability increasing protein and lipopolysaccharide(LPS)-binding protein.LPS binding properties and effects on LPS-mediated cell activation[J].J Biol Chem,1994,269(26):17411-17416.
[2] 王松明,顾招兵,朱雅新,等.蛋白质定向进化技术概述[J].中国饲料,2017(14):15-19.
[3] Moore J C,Jin H M,Kuchner O,et al.Strategies for theinvitroevolution of protein function:enzyme evolution by random recombination of improved sequences[J].J Mol Biol,1997,272(3):336-347.
[4] Stemmer W P.Rapid evolution of a protein in vitro by DNA shuffling[J].Nature,1994,370(6488):389-391.
[5] 李晶琴,李慎涛,温铭杰,等.人杀菌通透性增强蛋白功能性N端片段在酵母细胞表面的展示和鉴定[J].中国免疫学杂志,2007,23(8):738-742.
[6] Lin L,Fu C G,Huang W Q.Improving the activity of the endoglucanase,Cel8M from Escherichia coli by error-prone PCR[J].Enzyme Microb Technol,2016,86:52-58.
[7] 聂简琪,陈阿娜,刘秀霞,等.普鲁兰酶突变体文库高通量筛选方法的建立及应用[J].食品与生物技术学报,2016,35(9):993-1000.
[8] 李璟,童晋,罗明银,等.枯草芽孢杆菌脂肪酶基因lipaseA突变文库构建及其生物柴油转酯研究[J].浙江农业学报,2016,28(5):864-869.
[9] 乔超超,王新侠,李鹤宾,等.Pseudoalteromonas carrageenovora芳香基硫酸酯酶突变文库热稳定性提高突变体的筛选及鉴定[J].食品科学,2017,38(10):18-23.
[10] 陈晓穗,汪保安,王琰.错配PCR致突变的实验条件研究[J].第二军医大学学报,2003,24(3):307-310.
[11] Miyazaki K,Arnold F H.Exploring nonnatural evolutionary pathways by saturation mutagenesis:rapid improvement of protein function[J].J Mol Evol,1999,49(6):716-720.
[12] 周佳海,陈海宝.一种简便的DNA改组(DNA shuffling)操作程序[J].生物化学与生物物理进展,2000,27(6):655-657.
[13] 马玉成,朱涛,姜玉新,等.尘螨Ⅱ类变应原Der f2和Der p2的DNA改组及生物信息学分析[J].基础医学与临床,2012,32(6):634-638.
[14] 张凯,蔡恒,汪晨.通过DNA改组技术定向进化赖氨酸脱羧酶基因cadA和ldc[J].生物加工过程,2015,13(5):20-25.
[15] 金庆超,沈娜,杨郁,等.spy1的DNA改组提高普那霉素的产量[J].中国抗生素杂志,2015,40(3):178-191.
ConstructionandidentificationofgenemutationlibraryoftheN-terminalfragmentsofhumanbactericidal/permeabilityincreasingproteins
Li Jingqin1,Kong Qingli2,An Yunqing2*
(1.DepartmentofMedicalLaboratoryScience,YanjingMedicalCollege,CapitalMedicalUniversity,Beijing101300,China;2.DepartmentofImmunology,SchoolofBasicMedicalSciences,CapitalMedicalUniversity,Beijing100069,China)
ObjectiveTo construct and identify a gene mutation library of the N-terminal fragments of human bactericidal/permeability increasing proteins (BPI23) for enhancing its activity binding lipopolysaccharides (LPS) by error-prone polymerase chain reaction (PCR) and DNA shuffling method.MethodsError-prone PCR was used to obtain random mutant fragments ofBPI600by adding different concentrations of Mg2+and Mn2+.Random mutation rate was calculated by DNA sequencing and genetic comparison with wild-typeBPI600.The product of error-prone PCR was further mutated via DNA shuffling.The reassembled product was cloned into pYD1 vector and transformed intoE.coliXL10-Gold.Ten clones were randomly picked out,digested and identified by restriction enzymeHindⅢ andXhoⅠ.The positive clones were identified by DNA sequencing.ResultsDNA sequencing result showed that the random mutation rate was 2.3% by error-prone PCR.After DNA shuffling,6 positive clones includingBPI600were identified in the 10 random clones selected from LB culture medium with ampicillin.Among them,4 had 6,9,11 and 14 mutations,respectively;1 had 10 mutations and 1 deletion determined by DNA sequencing.By amino acid comparison,only 2 DNA mutants were able to encode a full-length mutant BPI23protein,with 4 and 14 amino acid mutations respectively.ConclusionA library of BPI23mutants was constructed with a size of 2×105.The results laid the foundation for the study of its activity binding LPS.
human bactericidal/permeability increasing protein;directed evolution;error-prone polymerase chain reaction(PCR);DNA shuffling
*Corresponding author,E-mail:anyunq@ccmu.edu.cn
时间:2017-12-13 21∶09
http://kns.cnki.net/kcms/detail/11.3662.R.20171213.2109.026.html
10.3969/j.issn.1006-7795.2017.06.024]
Q78
2017-10-16)
编辑 陈瑞芳
