水貂冠状病毒中国分离株全基因序列测定及分析
2017-12-06王楷宬庄青叶侯广宇
王楷宬,庄青叶,邱 源,侯广宇
(中国动物卫生与流行病学中心,山东青岛 266032)
水貂冠状病毒中国分离株全基因序列测定及分析
王楷宬,庄青叶,邱 源,侯广宇
(中国动物卫生与流行病学中心,山东青岛 266032)
为分析水貂冠状病毒中国分离株的遗传特征,采用高通量测序方法测定从山东省青岛市某养殖场分离的水貂冠状病毒基因组,首次报道了水貂冠状病毒中国分离株的全基因序列,并对其进行特性分析与比较基因组研究.结果显示:该病毒全基因组大小为 28 924 bp,具有与其他冠状病毒相似的基因组结构和基因顺序;5´端具有帽子结构,3´端具有poly(A)尾.该分离株与19株代表性毒株的全基因组和N基因进化和同源性分析都证实,该分离株与美国分离株的同源性较高,与其余2株水貂冠状病毒和2株雪貂冠状病毒同属于冠状病毒的一个新种.
水貂冠状病毒;中国分离株;基因组分析;高通量测序
由冠状病毒感染引起的水貂腹泻是除水貂细小病毒性肠炎外导致水貂腹泻的又一重要病毒性传染病,又称水貂流行性卡他性胃肠炎[1].病貂表现为:食欲不振、呕吐、精神萎靡、口渴、腹泻,排灰白色、绿色乃至粉黄色黏液状稀便,有的排黑红色、没有明显套管样稀便;鼻镜干燥,被毛欠光泽,消瘦,皮肤缺乏弹性;体温一般不高.腹泻严重的病貂,往往因脱水而发生自体中毒而死.该病最早流行于美国犹他州[1].1987年我国从北美进口种貂时带入本病[2].
冠状病毒是1968年由一位叫泰瑞的科学家发现的,因其形态在电子显微镜下,外膜呈日冕状或皇冠状而得名[3].冠状病毒粒子较大,形态多样,有囊膜;基因组为线状、单股、正链RNA分子;囊膜上镶嵌着许多一端较粗的膜粒(Peplomer),形成王冠状结构.每个膜粒由一个大的三聚体糖蛋白组成.该蛋白称为纤突蛋白(Spike)或S蛋白.S蛋白能够与宿主细胞受体结合,并介导病毒和宿主细胞的融合,也是自然感染过程中诱导宿主产生中和抗体的主要抗原成分.S蛋白中有些表位变异较快,可促使病毒逃避宿主的获得性免疫反应[3].
水貂冠状病毒的体外分离培养较为困难.有研究者曾试用10多种细胞进行分离培养,最后用貂肺细胞(ML)才获得成功[4].我国虽有水貂冠状病毒分离或鉴定的报道,但迄今尚未发表该病毒中国分离株的完整基因序列.为揭示水貂冠状病毒中国分离株的全基因序列特性,采用高通量测序方法,测定自山东省青岛市某养殖场分离的水貂冠状病毒基因组序列,并对其进行特性分析与比较基因组研究.
1 材料和方法
1.1 材料和仪器
Ion PGM Template OT2 400 Kit、Ion PGM Sequencing 400 Kit V2、E-Gel SizeSelect 2%Agarose、Ion 316 Chip Kit V2、Dynabeads MyOne Streptavidin C1 Beads、Qubit核酸浓度测定仪及配套试剂(Life technologies公司);超净工作台(美国Forma Scientific);移液器(Eppendorf);孵化器(德州诚信孵化设备有限公司);高速台式离心机(德国Heraeus Biofuge primoR);PCR扩增仪(Perkin Elmeter Gen Amp PCR System 9600).高性能计算平台:Dell T630塔式服务器,具有2颗Intel(R)Xeon(R)CPU E5-2620 v3 @2.40GHz,内存264 G,存储23 T,操作系统版本CentOS Linux release 7.1.1503(Core).
1.2 核酸提取
自山东省青岛市某养貂场2016年春季采集的粪便样品中,检测到水貂冠状病毒(Mink/China/1/2016)[5].将该份样品充分混匀后,经高速离心和过滤,去除大分子物质和细菌,使用QIAamp Viral RNA Mini Kit(Qiagen,德国)提取病毒核酸.
1.3 高通量测序(NGS)文库构建
对提取的病毒核酸,采用本中心已建立的方法[5]进行NGS文库构建.具体方法为:在8 µL病毒RNA中,加入1 µL 100 µmol/L引物A15N6和2 µL无核酸酶的水进行混合,72 ℃ 5 min后,将RNA引物混合物放在冰上孵育至少3 min;在混合物中加入 4 µL 5Xfirst-strand buffer、1 µL dNTP(100 μmol/L)、2 µL DTT(0.1 mol/L)、1 μL RNaseOUT™ Recombinant Ribonuclease Inhibitor(40 U/µL) 和 1 μL SuperScript® III Reverse Transcriptase (200 U/μL) (Invitrogen,USA),25 ℃反应 15 min,42 ℃反应 30 min,70 ℃ 15 min,终止反应;在反应产物中再加入1 µL RNase H(TaKaRa,Japan),37 ℃反应20 min;采用DynaMag ™ -2 Magnet 和 Agencourt® AMPure® XP Reagent(Beckman Coulter,USA)纯化合成第一链cDNA;在纯化的第一链cDNA中,加入引物B15N6,70 ℃ 5 min,再加入1 μL Klenow fragment (5 U) (NEB,USA),5 μL 10XNEBuffer 2、2 μL dNTP(100 μmol/L) 和 1 μL DTT(0.1 mol/L),37 ℃ 反应30 min;最后采用1XPhusion High-Fidelity Buffer,10 μmol/L 引 物 A30和 B30( 表1),0.5 U Phusion High-Fidelity DNA Polymerase(NEB,USA),进行PCR扩增.将PCR产物,采用E-Gel® SizeSelect™ Agarose Gel进行电泳,筛选和回收约450 bp的扩增产物,将其作为NGS文库.

表1 引物与引物序列
1.4 NGS测序
将NGS文库稀释为2.6X1011mol/L,应用Ion PGM Template OT2 400 Kit,对DNA文库进行测序前的样品处理.将处理后的样品加样至Ion 316芯片,置于PGM测序仪进行测序.
1.5 测序数据分析
对NGS测序数据经质控后,采用CLC genomics workbench 8.5.1(Qiagen,Germany),将测序得到的reads,参考GenBank中已发表的水貂冠状病毒WD1133全基因序列[6](GenBank登录号为HM245926)进行mapping 拼接,并分析拼接完成的基因组中的ORF;将注释后的序列提交GenBank.
1.6 基因组特性分析
采用MEGA 6.0[7]将Mink/China/1/2016的全基因与N基因,分别与GenBank中冠状病毒科的代表毒株(表2)全基因与N基因进行比对和进化分析,并对这些毒株的全基因进行同源性分析.

表2 基因分析所使用的冠状病毒毒株
2 结果与分析
2.1 基因组解析
芯片上样率为73%.测序文库得到质量较高的测序数据.CLC genomics workbench 8.5.1拼接得到水貂冠状病毒中国分离株的全基因大小为28 924 bp,GenBank登录号为 MF113046.Mink/China/1/2016基因组具有与其他冠状病毒相似的结构和基因顺序:5´端具有帽子结构,3´端具有A尾;基因组(图1)中主要包含ORF1a/1b、纤突蛋白(Spike protein,S)、3c、囊膜蛋白(Envelope protein,E)、膜蛋白(Membrane protein,M)、核衣壳蛋白(Nucleocapsid protein,N),以及其他附属编码非结构蛋白的基因(ORF7a、3x和7b);ORF1a和ORF1b相互重叠42 bp,其长度分别是12 030和8 034 bp;在ORF1a~ORF1b重叠区域(12 262~12 304)形成一个基因三级结构,可由其引起核糖体阅读框移位;与其他冠状病毒相似,核糖体移动的滑动位点为UUUAAAC;在非转录酶基因(3x和7b 除外)的上游均具有转录调控序列(CTAAAC),其是不间断合成亚基因组RNA所必须的.

图1 Mink/China/1/2016基因组结构
2.2 基因组比较
Mink/China/1/2016与19株代表性毒株(表2)的全基因组进化分析结果见图2,保守基因(N基因)的进化分析见图3,各毒株之间的全基因同源性分析见图4.分析结果显示,Mink/China/1/2016与已报道全基因的2株水貂冠状病毒(WD1127和WD1133)亲缘关系最近,基因组同源性分别为92.7%和93.8%,且此3株病毒在全基因和N基因的进化树中都归为一簇,并与2株雪貂冠状病毒(NL-2010和4370-7)明显归为一个与Alphacoronavirus 1并列的小分支.按照之前的报道[6],将此分支暂时命名为Alphacoronavirus 2(尚未被病毒分类委员会认可).在全基因和N基因的进化树中,各病毒的进化关系相似.
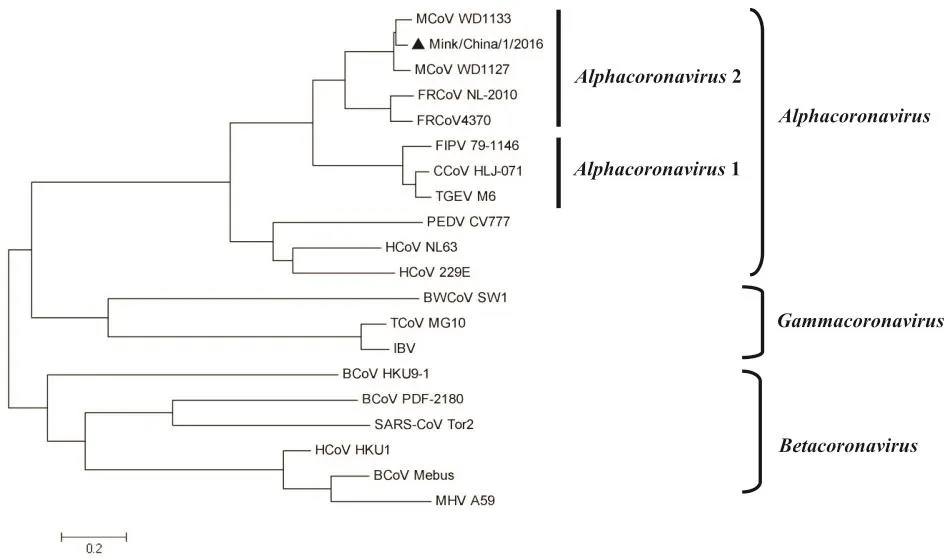
图2 冠状病毒全基因进化分析
3 讨论
我国水貂养殖业始于1956年.有报道称,2005年全国约有水貂饲养场户5 000 多家,种貂存栏250万只,年产水貂皮800万张[8].近几年,我国水貂饲养数量迅速增长,已经成为世界上较大的毛皮生产国和成品出口国.水貂消化道疾病往往导致水貂死亡或皮毛品质下降,造成较大经济损失.
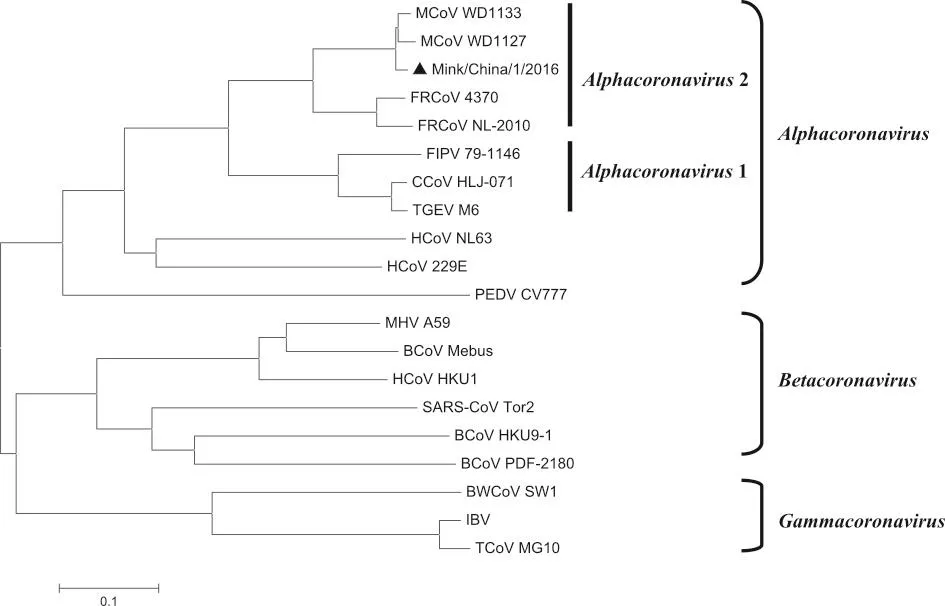
图3 冠状病毒全基因进化分析

图4 Mink/China/1/2016 与冠状病毒代表毒株的同源性分析
我国对水貂疫病的研究较少,主要针对犬瘟热病毒、水貂细小病毒和新城疫病毒感染等[9-10].而对水貂冠状病毒,尤其是该病毒的分子生物学特性研究较少.
Anastasia等[6]于2011年首次报道了2株分离自美国的水貂冠状病毒全基因序列,之后再无其他水貂冠状病毒全基因的报道.我国早在20世纪80年代就发生了由水貂冠状病毒引起的水貂流行性卡他性胃肠炎,但一直未对水貂冠状病毒流行毒株的分子生物学特性进行研究.本研究采用高通量测序方法,解析1株分离自青岛市的水貂冠状病毒(Mink/China/1/2016)全基因.此属该病毒中国分离株基因组的首次报道,也是世界上第3个该病毒全基因的报道.
对Mink/China/1/2016的全基因分析发现,该病毒基因组与2011年报道的2株美国分离株基因组极为相似,同源性分别达92.7%和93.8%,但由于3株毒株基因组中,有一些与ORF翻译起始和终止位点发生了变化,同一ORF在3株毒株中的大小存在差异,其翻译的蛋白大小也随之发生了改变.此变化是否会引起病毒致病力的改变有待进一步解析.我国分离株与美国分离株差异不大.这也可能与该病原由北美进口种貂带入有关.N基因是冠状病毒保守的结构蛋白.本研究选用此基因进行进化分析,经与文献报道[6]对比分析发现,该毒株Mink/China/1/2016与其余2株水貂冠状病毒和2株雪貂冠状病毒同属于冠状病毒的一个新种(Alphacoronavirus 2).本分析结果进一步验证了Anastasia等[6]的研究结论,为此新种的认可增加了科学依据.
[1] GORHAM J R,EVERMANN J F,WARD A,et al.Detection of coronavirus-like particles from mink with epizootic catarrhal gastroenteritis[J]. Canadian journal of veterinary research,1990,54(3):383-384.
[2]韩慧民,刘维全,杨盛华,等. 水貂冠状病毒性肠炎研究初报[J]. 兽医大学学报,1988,8(2):169.
[3]陈继明,马洪超,陆承平. 兽医微生物及所致传染病[M]. 2版. 北京:中国农业出版社,2015.
[4]韩慧民,刘维全,杨盛华,等. 水貂肠道冠状病毒的分离鉴定[J]. 兽医大学学报,1992,12(1):65.
[5] QIU Y,CHEN J M,WANG T,et al. Detection of viromes of RNA viruses using the next generation sequencing libraries prepared by three methods[J]. Virus research,2017,237:22-26.
[6] VLASOVA A N,HALPIN R,WANG S,et al. Molecular characterization of a new species in the genus Alphacoronavirus associated with mink epizootic catarrhal gastroenteritis[J].Journal of general virology,2011,92(Pt 6):1369-1379.
[7]TAMURA K,STECHER G,PETERSON D,et al.MEGA6:molecular evolutionary genetics analysis version 6.0[J]. Molecular Biology Evolution,2013,30(12):2725-2729.
[8]张志明. 从中国与丹麦、美国水貂养殖现状比较看中国水貂产业化发展方向[J]. 特种经济动植物,2005,8(9):2-5
[9] WANG Z,WU W,HU B,et al. Molecular epidemiology of Aleutian mink disease virus in China[J]. Virus research,2014,184:14-19.
[10] ZHAO P,SUN L,SUN X,et al. Newcastle disease virus from domestic mink,China,2014[J]. Veterinary microbiology,2017,198:104-107.
(责任编辑:朱迪国)
Whole Genome Sequencing and Analysis on Mink Coronavirus Isolated from China
Wang Kaicheng,Zhuang Qingye,Qiu Yuan,Hou Guangyu
(China Animal Health and Epidemiology Center,Qingdao,Shandong 266032)
To analyze the genetic characteristics of Mink coronavirus isolated from China,the genome of Mink coronavirus isolated from a farm of Qingdao city was determined by high throughput sequencing. The full-length genome sequence of the Mink coronavirus Chinese isolate was reported for the first time. The genomic sequence of the isolate was analyzed and compared with reference coronavirus. The results showed that the full-length of the isolate was 28 924 bp,which possessed genomic structure and gene order similar to the reference coronaviruses. The 5' end had a hat structure,and the 3' end had poly(A)tail. The genome and N gene evolution and homology analysis between the isolate and other 19 representative strains showed that the isolate had a high homology with the American isolate,and was a new species belonging to the coronavirus with the other 2 Mink coronaviruses and 2 ferret coronaviruses.
Mink coronavirus;Chinese isolates;genome analysis;high-throughput sequencing
S851.3
B
1005-944X(2017)12-0088-04
10.3969/j.issn.1005-944X.2017.12.025
国家重点研发计划项目(2017YFC1200500);中国动物卫生与流行病学中心创新基金(2015IF-0004FF)
