儿童囊性纤维化5例临床及影像表现
2017-11-29钟玉敏王诗渝
孙 燕 钟玉敏 朱 铭 王诗渝 王 剑 张 皓 张 磊 邵 虹
1.上海交通大学医学院附属上海儿童医学中心(上海 200127); 2.上海交通大学医学院(上海 200025)
儿童囊性纤维化5例临床及影像表现
孙 燕1钟玉敏1朱 铭1王诗渝2王 剑1张 皓1张 磊1邵 虹1
1.上海交通大学医学院附属上海儿童医学中心(上海 200127); 2.上海交通大学医学院(上海 200025)
目的探讨儿童囊性纤维化的临床及影像表现。方法回顾分析5例囊性纤维化患儿的临床及影像资料。结果5例患儿中,男3例、女2例,中位年龄6岁(2~13岁)。4例表现为反复咳嗽、咳痰,伴或不伴发热、气促;胸部CT均提示肺炎、支气管扩张伴支气管壁增厚、黏液嵌塞;鼻窦CT提示鼻窦炎、窦腔内密度异常增高,额窦发育不全,其中3例痰培养示铜绿假单胞菌感染。1例表现肝功能异常1年,腹部MRI示肝硬化、门脉周围组织T1WI呈高信号,胸部CT提示小气道阻塞造成空气潴留、支气管扩张伴黏液嵌塞。基因检测5例均有基因突变,共发现7个CFTR突变基因,其中2个为新发突变。结论囊性纤维化的影像表现具有一定特征,对临床诊断具有重要提示意义;中国人囊性纤维化基因突变位点与高加索人有一定差异。
囊性纤维化; X线计算机体层摄影术; 磁共振成像; 囊性纤维化穿膜传导调节因子
囊性纤维化(cystic fibrosis,CF)是高加索人常见的常染色体隐性遗传疾病,是由囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)基因突变引起[1]。CF在欧洲、北美洲、澳大利亚发病率达1/1 800~5 000[2],亚洲和非洲少见。日本总结1951-1993年间发生于日本的104例CF病人,认为CF在日本的发病率约1/350 000[3]。自1974年至今,中国共报道CF约55例[4-7],其中43例来自内地。现将确诊的5例CF病例报告如下,并结合国内外文献进行分析,以提高对此病的临床和影像认识。
1 临床资料
上海交通大学医学院附属上海儿童医学中心2015年11月-2016年12月确诊为CF患儿共5例。男3例、女2例,年龄2~13岁,中位年龄6岁。5例患儿中,4例因反复咳嗽、咳痰,伴或不伴发热、气促就诊;均有血白细胞、中性粒细胞及C反应蛋白升高,其中3例痰培养提示铜绿假单胞菌感染,1例肺功能提示轻到中度以阻塞为主的混合性病变。另1例因肝功能异常、皮肤巩膜黄染1年就诊,肝功能检测肝酶明显升高(表1)。5例患儿均无家族史,父母非近亲结婚。
5例患儿均行胸部CT检查,4例行鼻窦CT检查。1例患儿行腹部MR检查。例1、3行常规胸部X线检查。
胸部CT检查提示不同程度支气管扩张(印戒征)伴支气管壁增厚、黏液嵌塞(小叶中心结节、树芽征)、感染,其中1例局部肺野透亮度增高,1例胸片显示两膈低平,均提示小气道梗阻造成空气潴留。见图1~3。
4例鼻窦CT(图1、2)提示额窦发育不全、鼻窦炎、窦腔内CT值明显增高,达45~54 HU,其中1例上颌窦内侧壁向内侧膨出(图1)。

图1 例1患儿影像图

图2 例3患儿胸部及鼻窦CT

图3 例5患儿影像图
5例中1例腹部MR T1WI序列提示肝硬化、再生结节形成、门脉高压,肝内多发低信号分隔,门脉周围组织增厚,呈特征性T1WI高信号(图3)。
经家长知情同意,采集2 mL外周血标本,对CFTR基因的27个外显子和空白序列通过聚合酶链反应(polymerase chain reaction,PCR)方法进行扩增。利用Agilent SureSelect方法外显子捕获,Illumina测序平台进行高通量测序,测序数据经NextGENe®软件匹配分析后,用Ingenuity在线软件系统进行变异过筛及解释,候选变异经Sanger测序验证。5例患儿均存在基因突变,共检测到7个CFTR突变基因,其中2个为新发现变异。见表1。
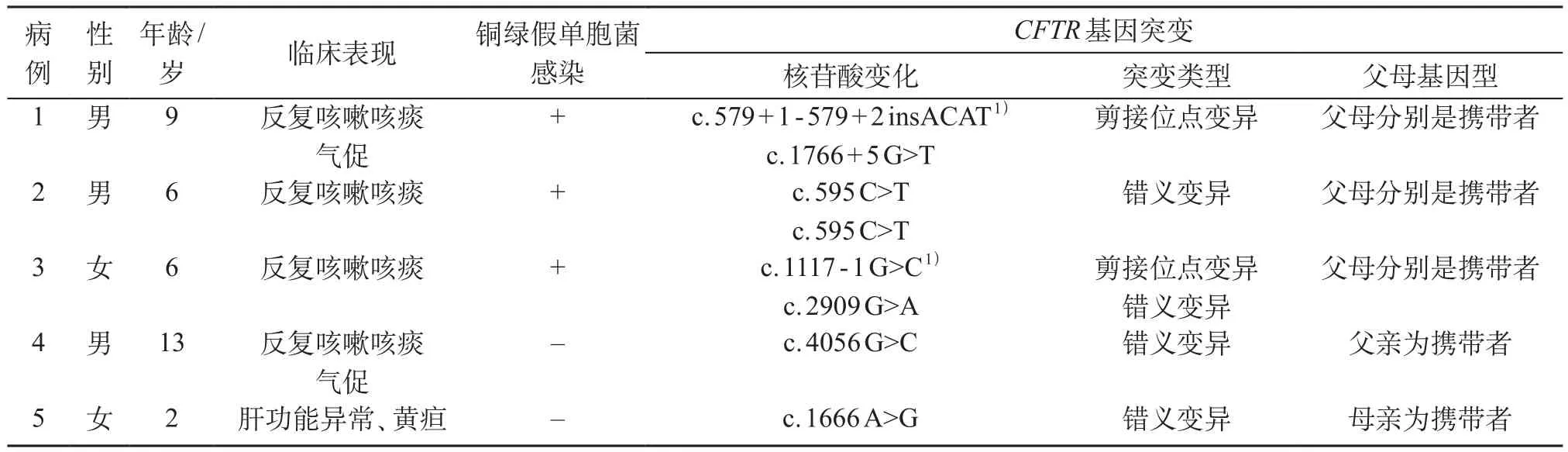
表1 5例患儿临床及基因检测结果
2 讨论
CF是高加索人群常见的致死性常染色体隐性遗传性疾病,以往认为在中国的发病率非常低,自1974年起,共报道约55例[4-7],其中近半数病例来自近3年。随着了解的深入并结合新近文献[4,5]提示,过去CF在中国的发病率很可能由于认识不足、检测技术的不完善被低估,本组患儿从首次就诊到确诊经历1~13年。准确、及早诊断CF对于患儿的个体化治疗有十分重要的意义[8]。
CF是由7号染色体长臂上的基因突变引起,该基因编码产物囊性纤维化跨膜传导调节因子CFTR是一种cAMP调节的氯离子通道蛋白,位于外分泌腺上皮细胞尖端。基因突变导致该蛋白合成、翻译异常及功能丧失,使外分泌腺上皮细胞对氯离子的通透性减低,对钠离子吸收增加,引起细胞内高渗状态,分泌液黏稠,可累及全身多个器官和系统[1,9]。
到目前为止,已发现超过2 000个CFTR基因突变。欧洲和北美最常见的基因突变是ΔF508[10],占一半以上,该基因在我国以及日本、韩国均未见报道。本组7个基因突变均不属于高加索人常见的突变,这与国内学者的报道一致[4,5],可见中国人与高加索人基因突变谱有相当的差异。c.1117-1G>C,c.579+1-579+2insACAT未在HGMD与ExAC等数据库收录,为新发现的变异,经Alamut功能软件预测,可能严重影响mRNA的编辑加工,属于“可能致病性变异”。c.1666A>G在中国人CF有一定的出现概率,近年两篇文章里共报道4例,c.1766+5G>T有1例报道,c.2909G>A 有2例报道[4,5]。
CF临床表现差异较大,可累及汗腺、肺、肝、胰腺、小肠、男性输精管等[1,11]。黏液-纤毛清除系统受损导致CF患者反复肺部感染[12],是其致死的主要原因。铜绿假单胞菌感染最常见[13,14]。本组4例肺部感染中3例为该菌感染。肺受累的早期征象是小气道阻塞造成空气潴留,胸片、胸部CT都可提示,如胸片显示两膈低平,胸部CT局部肺野密度减低。胸部CT的一个特征性表现为支气管扩张(印戒征)、支气管管壁增厚。1例以肝硬化为主要表现的患儿虽无呼吸道症状,但胸部CT证实了支气管扩张的存在,对全面了解CF病情及预防感染起到一定作用。胸部CT的另一特征性表现是扩张的支气管内的黏液嵌塞,这与黏液增多黏稠及排解不畅有关。因截面的不同,黏液嵌塞可以表现为树芽征、小叶中心结节、扩张支气管内的条柱状密度增高影,多平面重建的应用可将气道内的黏液栓更为直观地显示出来。胸部CT显示黏液嵌塞及其严重的1例,临床医师对其进行支气管肺泡灌洗时冲洗出大量脓液,显示胸部CT对临床诊治具有参考意义。
在儿科中支气管扩张并不常见,这一征象需要与其他感染、遗传性疾病鉴别。原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)是一种少见的常染色体隐性遗传病,患者纤毛超微结构异常导致黏液纤毛转运障碍,临床表现也是反复呼吸道感染、支气管扩张、鼻窦炎,鉴别诊断依靠支气管镜取支气管黏膜上皮在电镜下观察纤毛数目及结构异常。文献报道CF的支气管扩张通常以上肺野为著,甚至可以作为与PCD鉴别的参考依据[15],但在本组患者中表现均不明显,有待今后通过更多的病例进一步观察。Kartagener综合征以内脏反位、支气管扩张、鼻窦炎为三联征,行胸腹部检查观察内脏反位可供鉴别。马凡综合征肺部可有支气管扩张,无眼部症状、蜘蛛指(趾)、超声心动图无瓣膜病变可排除该病。
鼻窦炎是CF累及上呼吸道的另一表现[16]。本组4例鼻窦CT均显示额窦未气化,其中例3上颌窦、筛窦几乎完全被分泌物填充,且分泌物CT值高达47~54 HU,远远高于普通鼻窦炎(一般不超过15 HU),证明其黏稠性高,与CF的发病机制相符。文献中提到额窦发育不全及上颌窦、筛窦腔内分泌物累及范围超过75%是CF的特征性改变[17],本组病例鼻窦影像表现典型。
只有33%的CF发生肝胆病变,但其致死率仅次于CF的肺部感染和移植并发症,居第三位[18]。组织学上将肝脏CF病理改变分为3类:肝脂肪沉积,局灶性胆管纤维化,肝硬化,诊断依靠临床评估、生化检测、影像评估[19]。影像检查方法中超声应用最广泛,其特征是局灶性胆管纤维化,表现为门脉周围组织增厚(>2 mm)、回声增高[18]。磁共振成像联合MRCP优势明显,不仅可显示肝脏脂肪沉积的程度、肝硬化伴再生结节形成、门脉周围组织增厚,还可显示肝内外胆管的异常。例5 T1WI门脉周围组织呈特征性高信号,研究指出,这种高信号可被压脂序列抑制,提示为脂肪沉积[20]。这在病理学上与其他研究者在CF患者门脉周围组织内发现含脂肪的细胞一致[21]。这一特点可用于与朗格汉斯组织细胞增生症(Langerhans cells hyperplasia,LCH)鉴别。LCH肝脏受累时门脉周围组织增厚,T1WI呈低信号。在实际影像诊断中,可依此排除LCH,结合其胸部CT表现,考虑CF的可能。
汗液检测是CF重要而准确的检测手段,CF患儿汗液中氯离子浓度高于60 mmol/L[22]。然而汗液试验对操作要求很严格,操作上的微小错误会造成氯离子浓度错误而影响诊断。在国外,汗液试验也仅由获得认证的专业CF中心来进行。本组CF患儿未进行汗液试验,仅通过基因检测确诊,这是本研究的不足之处,也是国内很多医院CF研究和诊断受限的原因之一。本研究的另一不足之处是CF可累及多系统,但是本院CF患儿只涉及呼吸系统和/或肝脏,一方面与CF发病率低、病例数少有关,另一方面与中国CF表型是否有关有待丰富病例再进一步探究。
综上所述,CF在肺部、鼻窦、肝脏的影像表现具有特征性,包括小气道阻塞造成的空气潴留征象、支气管扩张伴支气管壁增厚、黏液嵌塞(小叶中心结节、条柱状密度增高影、树芽征)、鼻窦腔密度的异常增高、肝内门脉周围组织T1WI高信号等。由于认识不足、检测手段不完善可造成延误诊治,因此应该了解熟悉该病的临床和影像表现,以提供诊断线索。
[1]Elborn JS.Cystic fi brosis [J].Lancet,2016,10(2): 128-132.
[2]Haack A,Aragão GG,Novaes MR.Pathophysiology of cystic fi brosis and drugs used in associated digestive tract diseases.[J].World J Gastroenterol,2013,19(46): 8552-8561.
[3]Yamashiro Y,Shimizu T,Oguchi S,et al.The estimated incidence of cystic fibrosis in Japan.[J].J Pediatr Gastroenterol Nutr,1997,24(5): 544-547.
[4]Shen Y,Liu J,Zhong L,et al.Clinical phenotypes and genotypic spectrum of cystic fi brosis in Chinese children [J].J Pediatr,2016,171: 269-276.
[5]Liu Y,Wang L,Tian X,et al.Characterization of gene mutations and phenotypes of cystic fibrosis in Chinese patients.[J].Respirology,2015,20(2): 312-318.
[6]王苹莉,景继勇,沈华浩.20例中国人囊性纤维化回顾分析[J].中华儿科杂志,2008,46(8): 634-636.
[7]刘金荣,彭芸,赵宇红,等.中国儿童囊性纤维化二例临床特点及基因分析[J].中华儿科杂志,2012,50(11): 829-833.
[8]Bell SC,Boeck KD,Amaral MD.New pharmacological approaches for cystic fi brosis: promises,progress,pitfalls [J].Pharmacol Ther,2015,145: 19-34.
[9]Cutting GR.Cystic fibrosis genetics: from molecular understanding to clinical application [J].Nat Rev Genet,2015,16(1): 45-56.
[10]De Boeck K,Zolin A,Cuppens H,et al.The relative frequency of CFTR,mutation classes in European patients with cystic fi brosis [J].J Cyst Fibros,2014,13(4): 403-409.
[11]Mickle JE,Cutting GR.Genotype-phenotype relationships in cystic fi brosis.[J].Med Clin North Am,2000,84(3): 597-607.
[12]Hoegger MJ,Fischer AJ,Mcmenimen JD,et al.Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fi brosis [J].Science,2014,345(6198): 818-822.
[13]Knibbs LD,Johnson GR,Kidd TJ,et al.Viability ofPseudomonas aeruginosain cough aerosols generated by persons with cystic fibrosis.[J].Thorax,2014,69(8): 740-745.
[14]Cantin AM,Hartl D,Konstan MW,et al.Inflammation in cystic fi brosis lung disease: pathogenesis and therapy.[J].J Cyst Fibros,2015,14(4): 419-430.
[15]Kennedy MP,Noone PG,Leigh MW,et al.High-resolution CT of patients with primary ciliary dyskinesia.[J].AJR Am J Roentgenol,2012,188(5): 1232-1238.
[16]Chang EH.New insights into the pathogenesis of cystic fibrosis sinusitis [J].Int Forum Allergy Rhinol,2014,4(2):132-137.
[17]Mak GK,Henig NR.Sinus disease in cystic fi brosis [J].Clin Rev Allergy Immunol,2001,21(1): 51-63.
[18]Flass T,Narkewicz MR.Cirrhosis and other liver disease in cystic fi brosis [J].J Cyst Fibros,2012,12(2): 116-124.
[19]Parisi GF,Di Dio G,Franzonello C,et al.Liver disease in cystic fibrosis: an update.[J].Hepat Mon,2013,13(8):e11215.
[20]King LJ,Scurr ED,Murugan N,et al.Hepatobiliary and pancreatic manifestations of cystic fibrosis: MR imaging appearances.[J].Radiographics,2000,20(3): 767-777.
[21]Hultcrantz R,Mengarelli S,Strandvik B.Morphological findings in the liver of children with cystic fibrosis: a light and electron microscopical study [J].Hepatology,1986,6(5):881-889.
[22]Mckone EF,Velentgas P,Swenson AJ,et al.Association of sweat chloride concentration at time of diagnosis andCFTRgenotype with mortality and cystic fi brosis phenotype [J].J Cyst Fibros,2015,14(5): 580-586.
2017-05-05)
(本文编辑:梁 华 )
Clinical and radiological manifestations of 5 pediatric cases with cystic fibrosis
SUN Yan1,ZHONG Yumin1,ZHU Ming1,WANG Shiyu2,WANG Jian1,ZHANG Hao1,ZHANG Lei1,SHAO Hong1(1.Shanghai Jiaotong University Affiliated Shanghai Children’s Medical Center,Shanghai 200127,China; 2.Shanghai Jiaotong University School of Medicine,Shanghai 200025,China)
ObjectiveTo explore the clinical manifestations and radiological features of cystic fi brosis (CF) in children.MethodsThe clinical and radiographic data of 5 CF patients were retrospectively analyzed.ResultsAmong the 5 cases,there are 3 males and 2 females,aging from 2 to 13 years old (median age 6).Four of the fi ve cases had complaints of repeated productive cough with or without fever and short breath.Pseudomonas aeruginosawas positive in sputum culture of three cases.Chest CT showed pneumonia and bronchiectasis with peribronchial thickening and mucus plugging.Paranasal CT showed frontal sinus agenesis and sinusitis with sticky secretion.The other one of the 5 cases had a complaint of abnormal hepatic function.The abdominal MRI showed liver cirrhosis and high signal intensity in the periportal area on T1-weighted imaging.Chest CT showed air trapping from small airways obstruction and bronchiectasis with sputum plugging.Five recurrent and two novelCFTRmutations were identi fi ed in all of the 5 cases.ConclusionsThe radiographic fi ndings of CF are characteristic,and of great signi fi cance to the clinical diagnosis of CF.The gene mutations of CF in Chinese are different from those in Caucasians.
cystic fi brosis; X-ray computed tomography; magnetic resonance imaging; cystic fi brosis transmembrane conductance regulator
10.3969/j.issn.1000-3606.2017.11.009
邵虹 电子信箱:wyqrainbow@hotmail.com
