CYP27A1基因突变致脑黄腱瘤1例报告并文献复习
2017-11-29郭红梅
郭红梅 李 玫 金 玉 杨 光
南京医科大学附属儿童医院消化科(江苏南京 210008)
CYP27A1基因突变致脑黄腱瘤1例报告并文献复习
郭红梅 李 玫 金 玉 杨 光
南京医科大学附属儿童医院消化科(江苏南京 210008)
目的探讨固醇-27羟化酶(CYP27A1)基因变异引起的脑黄腱瘤患儿的临床特点,肝脏病理改变及预后。方法回顾分析1例CYP27A1基因变异引起的脑黄腱瘤患儿的临床特点,并复习相关文献。结果患儿,女,1月龄,表现为胆汁淤积、肝大、转氨酶升高,谷氨酸转移酶及总胆汁酸正常;病理检查提示肝内胆汁淤积、炎症细胞浸润,毛细胆管扩张及增生;基因检测示CYP27A1基因剪接位点c.1263+1G>A / c.1477-3C>G复合杂合变异,其中c.1477-3C>G为一新颖变异。结论婴儿期出现胆汁淤积、转氨酶升高、肝肿大,而谷氨酸转移酶及总胆汁酸正常或减低,需警惕胆汁酸合成障碍,应尽早完善基因检测,以早期诊断及治疗,改善预后。
胆汁淤积; 代谢缺陷; 先天性疾病
脑腱黄瘤病(OMIM#213700,cerbrotendinous xanthomatosis,CTX)是一种常染色体隐性遗传的脂质代谢性疾病,发病率约为1/70 000,中国人群发病率极低,其中女性发病率稍高于男性[1]。目前国内共报道17个家系19例CTX患者[2]。中国人 CTX的首发症状多为白内障和双侧跟腱部肿物,而后逐渐出现肌腱黄色瘤(跟腱最多见),尚无胆汁淤积征为首发表现的报道。现报告1例胆汁淤积征患儿,经基因检测发现固醇-27羟化酶(CYP27A1)基因杂合突变,诊断为CTX。
1 临床资料
患儿,女,1个月10天,因皮肤、巩膜黄染1月余入院,生后1周左右无明显诱因出现巩膜黄染,渐加重出现全身皮肤黄染。当地医院诊断为“新生儿黄疸”予茵栀黄口服液口服4天,效果欠佳,遂予蓝光治疗1天,患儿皮肤黄染无明显改变。病程中大便黄色糊状,带有白色奶瓣,尿色深,量可;谷氨酸氨基转移酶167 U/L,天冬氨酸氨基转移酶194 U/L,总胆红素187.7 μmol/L,直接胆红素90.3 μmol/L,间接胆红素97.4 μmol/L,总蛋白53.5 g/L,遂至南京医科大学附属儿童医院就诊,拟诊“胆汁淤积性肝病”收住院。病程中患儿精神反应可,无发热、咳嗽、呕吐、抽搐,食纳睡眠可;G1P1,足月剖宫产,出生体质量4.3 kg;父母体健,非近亲结婚。患儿入院后予保肝、退黄及对症支持治疗,完善相关检查。实验室检查:谷氨酸氨基转移酶202 U/L,天冬氨酸氨基转移酶248 U/L,总胆红素206.48 μmol/L,直接胆红素146.12 μmol/L,间接胆红素60.36 μmol/L, γ谷氨酰转肽酶(γ-GT)72 U/L,总蛋白52.8 g/L,白蛋白40.4 g/L,球蛋白12.4 g/L,胆汁酸23 μmol/L,总胆固醇4.66 mmol/L;肝胆核素显像符合胆道梗阻表现。完善术前准备后,在全麻下行剖腹后胆道造影术。术中见肝脏稍增大,术中胆道造影示胆管细,予冲洗胆道。术后病理报告:肝小叶结构不明显,肝细胞变性坏死融合,胆汁淤积、汇管区较多的慢性炎症细胞浸润,毛细胆管扩张及增生。术后复查谷氨酸氨基转移酶380 U/L,天冬氨酸氨基转移酶375 U/L,总胆红素216 μmol/L,直接胆红素171 μmol /L,间接胆红素42 μmol/L,γ-GT 57 U/L。进一步完善遗传代谢性肝病基因检查,并继续予口服复方甘草酸片保肝、熊去氧胆酸退黄疸治疗,门诊随诊。
经南京儿童医院伦理委员会批准,患儿家属的填写知情同意书后,抽取患儿及其父母外周静脉肝素抗凝血3 mL,送北京希望组生物科技有限公司进行肝脏遗传性疾病全基因检测。基因检测结果示患儿CYP27A1基因剪切位点c.1263+1G>A / c.1477-3C>G复合杂合突变。CYP27A1基因1263位后面的内含子的第一个碱基的G转变成了A(图1 a),该变异在 ExAC(http://exac.broadinstitute.org)普通人数据库东亚人群中的频率为 0.0001,这一变异为典型的剪接位点变异,可能导致蛋白功能丧失。已有文献报道在CTX患者中检测到该位点,体内实验证明该变异可引起蛋白功能丧失[3]。患儿母亲CYP27A1基因存在c.1263+1G>A 变异(图1c),患儿父亲CYP27A1基因无 c.1263+1G>A 变异(图1e)。患儿CYP27A1基因的 c.1477-3C>G 变异(图1 b)在 ExAC 普通人数据库东亚人群中的频率为0,该变异未在其父母中检出(图1d、f),为新发变异,剪接位点预测软件NetGene2(https://omictools.com/netgene2-tool)、NNSplice(https://omictools.com/nnsplice-tool)及FSPLICE(http://www.softberry.com)均预测该位点显著影响剪接作用,目前该变异尚未见文献报道。
患儿4月龄确诊脑黄腱瘤病,眼科会诊未发现白内障。头颅磁共振成像检查未见异常。开始口服鹅脱氧胆酸15 mg/(kg·d)。患儿5个月胆汁淤积消退,复查谷氨酸氨基转移酶63 U/L,天冬氨酸氨基转移酶48 U/L,总胆红素13.9 μmol/L,直接胆红素5.9 μmol/L,间接胆红素8 μmol/L,γ-GT 8 U/L。随访至患儿13月龄,未出现CTX其他临床表现。
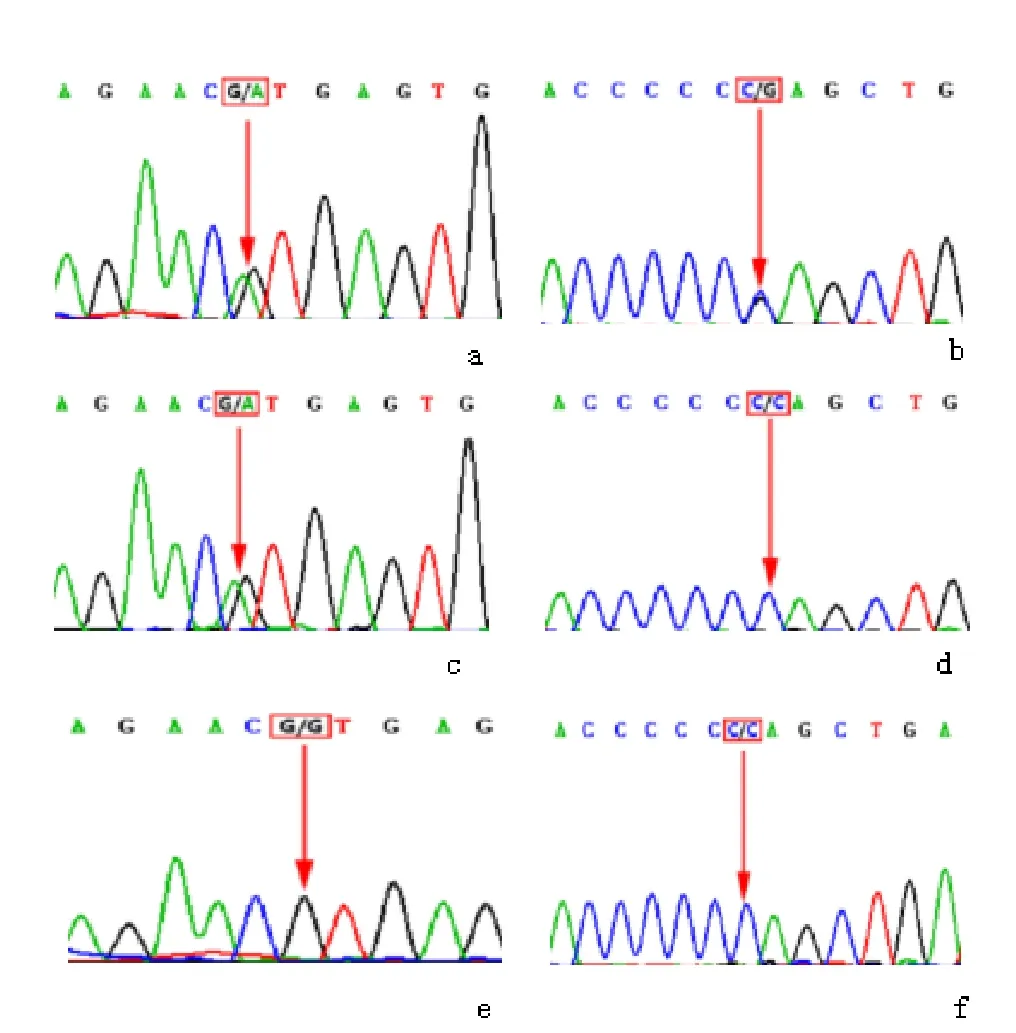
图1 CYP27A1基因分析结果
2 讨论
CTX致病基因CYP27A1位于2号染色体 q33-qter区,该基因突变导致固醇-27-羟化酶(CYP27A1)活性减低或消失,致使7α-羟基-4-胆甾烯-3-酮转化为胆汁酸和CDCA通路受损,胆汁酸和CDCA生成减少,对CYP27A1的负反馈作用减弱,从而使胆甾烷醇及胆汁醇生成增加,并异常堆积在晶状体、脑、肌腱、骨骼等组织中,出现相应的临床症状[4]。
各个年龄段CTX患者的表现各不相同。已有一些病例报道将新生儿胆汁淤积性肝病作为可能CTX的早期表现,长期不能解释的新生儿黄疸或胆汁淤积可作为提示CTX强力指证之一[5]。本例患儿生后1个月内发病,表现为胆汁淤积、肝大、转氨酶升高、γ-GT及总胆汁酸正常、胆固醇正常,合并脂溶性维生素A、D吸收不良。胆道造影无造影剂进入肠道而误认为胆道梗阻。全麻下行剖腹后胆道造影术,术中见肝脏稍增大,术中胆道造影提示胆管细,考虑胆管发育不良,予冲洗胆道。术后肝活检显示为胆汁淤积、汇管区较多的慢性炎症细胞浸润,毛细胆管扩张及增生。鉴于患儿胆汁淤积,但γ-GT及总胆汁酸正常的特点,需与进行性家族性肝内胆汁淤积病和先天性胆汁酸合成障碍相鉴别[6]。经遗传代谢性肝病的基因筛查,结果发现CYP27A1基因剪切位点c.1263+1G>A/c.1477-3C>G复合杂合突变,且c.1477-3C>G变异目前尚未见文献报道,为一新颖变异。儿童期腹泻和青少年期白内障也是CTX患者的早期表现。50%的CTX患者儿童期出现慢性难治性腹泻,但腹泻患儿胃肠道检查正常[7]。儿童期白内障合并长期腹泻对本病的早期诊断具有非常重要的指导价值。此外,精神运动发育迟缓,小脑和锥体束征同样比较常见[8]。成人表现为痉挛性瘫痪、痴呆、共济失调、抽搐,以及周围神经病变。成人中枢系统外表现包括黄色瘤病、早发动脉粥样硬化以及骨质疏松导致病理性骨折。本例患儿年龄小,发现早,尚未出现腹泻、白内障及精神运动发育迟缓,经口鹅脱氧胆酸治疗后,患儿5个月胆汁淤积消退,随访至今未出现CTX其他临床表现。
血浆胆甾烷醇及尿胆汁醇含量异常升高被认为是诊断CTX 的实验室指标,CTX患者血浆胆甾烷醇含量较正常水平高5~10倍,血浆胆汁醇含量可较正常水平高500~1 000倍[9]。CTX诊断最简单的方法是以电喷雾电离质谱法(ESI-MS)分析尿液,可以检测到大量的胆汁醇葡萄糖甘酸。CTX虽然是胆固醇代谢障碍引起异常沉积,但是血胆固醇水平可以不升高甚至降低。
早期检测和诊断CTX很关键,因为早期和长期治疗CTX可以改善神经系统症状,甚至逆转疾病进展。然而,目前普遍存在CTX自症状出现到明确诊断有明显的延迟。CTX诊断“金标准”是CYP27A1基因分析。CYP27A1基因共有9个外显子,突变已达 70 余种,其中 45%属错义突变,20%无义突变,18%剪接突变,14%缺失突变以及2%插入突变。超过 50%的突变发生于6号至8号外显子之间,14%位于2号外显子,14%位于4号外显子区域。中国人CYP27A1的1号、2号和5号外显子突变较多。本例患儿的变异均发生在内含子区,显著影响剪接作用。
CTX治疗包括替代治疗,外科手术和对症治疗。胆汁酸替代治疗的潜在机制可能是通过激活胆汁酸负反馈机制,外源性抑制胆汁酸生成。抑制α-羟基-4-胆甾烯-3-酮生成,从而使胆甾烷醇浓度恢复正常,防止胆甾烷醇在组织中蓄积。对比熊去氧胆酸或牛磺胆酸,鹅脱氧胆酸是治疗CTX患者神经和非神经症状的选择,胆酸对非神经系统症状的治疗有效[10]。但有研究发现,尽管鹅脱氧胆酸补充治疗可使血浆胆甾烷醇、尿胆汁醇水平恢复正常,但其并不能改善神经系统症状,经鹅脱氧胆酸 治疗1年后,60%患者的神经系统症状仍然出现进行性加重,并有20%患者死亡,提示CTX 患者生存率仅与疾病诊断年龄相关,早期诊断是改善 CTX 预后的最关键的因素[11]。
综上,对于婴儿期出现的胆汁淤积、转氨酶升高、肝肿大、γ-GT和总胆汁酸正常或降低的患儿,需警惕各种类型的胆汁酸合成障碍,应尽早完善基因检测,一旦确诊,早使用胆汁酸替代治疗,可以明显改善患儿的生存质量及预后。
[1]聂淑科,赵琪,张允建.脑腱黄瘤病[J].中华神经科杂志,2013,46(11): 784-786.
[2]张亮亮,王训,程楠,等.19例中国人脑腱黄瘤病的临床特点分析[J].第二军医大学学报,2016,37(9): 1180-1183.
[3]Garuti R,Lelli N,Barozzini M,et al.Cerebrotendinous xanthomatosis caused by two new mutations of the sterol-27-hydroxylasegene that disrupt mRNA splicing [J].J Lipid Res,1996,37(7): 1459-1467.
[4]Larson A,Weisfeld-Adams JD,Benke TA,et al.Cerebrotendinous xanthomatosis presenting with infantile spasms and intellectual disability [J].JIMD,2016,39(6): 891-895.
[5]Mignarri A,Gallus GN,Dotti MT,et al.A suspicion index for early diagnosis and treatmentof cerebrotendinous xanthomatosis [J].J Inherit Metab Dis,2014,37(3): 421-429.
[6]代东伶.先天性胆汁酸合成障碍[J].临床儿科杂志,2015,33(4): 301-305.
[7]Verrips A,van Engelen BG,Wevers RA,et al.Presence of diarrhea and absence of tendon xanthomas in patients with cerebrotendinous xanthomatosis [J].Arch Neurol,2000,57(4):520-524.
[8]Degos B,Nadjar Y,Amador Mdel M,et al.Natural history of cerebrotendinous xanthomatosis: a paediatric disease diagnosed in adulthood [J].Orphanet J Rare Dis,2016,11(1):1-4.
[9]姚甲瑞,黄德晖,吴卫平.脑腱黄瘤病及CYP27A1基因突变研究进展[J].实用医技杂志,2015,22(6): 614-616.
[10]Ginanneschi F,Mignarri A,Mondelli M,et al.Polyneuropathy in cerebrotendinous xanthomatosis and response to treatment with chenodeoxycholic acid [J].J Neurol,2013,260(1): 268-274.
[11]Pilo-de-la-Fuente B,Jimenez-Escrig A,Lorenzo JR,et al.Cerebrotendinous xanthomatosis in Spain: clinical,prognostic,and genetic survey [J].Eur J Eeurol,2011,18(10): 1203-1211.
2017-01-09)
(本文编辑:邹 强)
A case report of cerebrotendinous xanthomatosis with novel mutations inCYP27A1gene
GUO Hongmei,LI Mei,JIN Yu,YANG Guang(Children's Hospital Af fi liated to Nanjing Medical University,Nanjing 210008,Jiangsu,China)
ObjectiveTo discuss the clinical features,hepatic pathology,and prognosis of cerebrotendinous xanthomatosis in a child caused byCYP27A1mutation.MethodsClinical features of a child with cerebrotendinous xanthomatosis were retrospectively analyzed,and the related literatures viewed.ResultsThe child had different degrees of cholestasis,hepatomegaly,elevated transaminases,normal-glutamyl GGT(γ-GT) and normal total bile acid.The hepatic pathology showed intrahepatic cholestasis,in fl ammatory cell in fi ltration and expansion and hyperplasia of bile capillary.Gene testing found heterozygous mutations ofCYP27A1(c.1263+1G>A / c.1477-3C>G) in the child.The variant of c.1477-3C>G is a novel mutation.ConclusionsThe possibility of bile acid synthesis disorder should be considered when infants have cholestasis,elevated transaminase,hepatomegaly,and normal or reduced γ-GT and total bile acid.Gene testing should be used for early diagnosis,treatment to improve prognosis.
cholestasis; metabolism de fi ciency; congenital disease
10.3969/j.issn.1000-3606.2017.11.010
国家临床重点专科建设项目-小儿消化专业资助项目(No.2011873)
杨光 电子信箱:gyangnj@qq.com
