先天性纯红细胞再生障碍性贫血患儿临床表现及基因检测
2017-11-29石玉梅
石玉梅 马 杰
单县海吉亚医院儿科( 山东单县 274300)
先天性纯红细胞再生障碍性贫血患儿临床表现及基因检测
石玉梅 马 杰
单县海吉亚医院儿科( 山东单县 274300)
目的探讨先天性纯红细胞再生障碍性贫血(DBA)的临床特点及其致病基因。方法回顾分析2例DBA患儿的临床资料以及基因检测结果,同时复习相关文献。结果两例患儿均为女性,分别为3月龄和4月龄;均以面色苍黄就诊。血常规红细胞偏低、血红蛋白低、网织红细胞计数低;血清铁及铁蛋白均升高;红细胞脆性实验未见异常。骨髓细胞学均提示髓象幼红细胞罕见。基因检测,例1的RPS19存在c.91C>T(p.P31S)杂合突变,其父母未见突变,该突变为新发突变,经验证为致病基因;例2的RPL5基因存在c.472_473del缺失突变(p.K158fs),为已知致病基因。结论DBA患儿多在出生早期发病,临床表现为红系缺乏,RPS19及RPL5基因突变较常见,相关基因检测有利于早期诊断;c.91C>T(p.P31S)杂合突变为未见报道的新突变。
先天性纯红细胞再生障碍性贫血; 临床特点; 基因检测
先天性纯红细胞再生障碍性贫血又称Diamond-Blackfan贫血(Diamond-Blackfan anemia,DBA;MIM 105650)是一种先天性骨髓衰竭性疾病[1],因Diamond和Blackfan于1938年首先报道而得名。其血液学特点为巨幼红细胞性贫血,网织红细胞数减少,骨髓增生较活跃,粒系、巨核系细胞增生正常,而红系细胞明显缺乏[2]。欧美国家DBA的发病率为0.05/万~0.07/万[3],国内暂没有DBA发病率的统计结果。一种编码核糖体小亚基蛋白19基因(RPS19)首先被证实与DBA发病相关,随后研究人员陆续发现与DBA相关的其他核糖体蛋白基因,包括RPS17、RPL5、RPS24、RPS7等[4-6]。现回顾分析2例DBA患儿的临床资料,并结合相关文献进行复习,探讨DBA的临床表现及基因变异特点。
1 临床资料
例1,女,3月龄,因“面色苍黄近1个月”来单县海吉亚医院儿科就诊。无发热、呕吐、抽搐,大小便未见异常。入院后体格检查:贫血面容,神志清楚,全身皮肤未见皮疹;双侧呼吸对称,心律齐,心音有力,各听诊区未闻及杂音。实验室检查:血常规红细胞1.24×1012/L,白细胞4.28×109/L,血小板448×109/L,血红蛋白46 g/L,网织红细胞计数4.1×109/L,红细胞平均体积108.4 fL(参考值75~121 fL),红细胞平均血红蛋白量34.5 pg(25~35 pg),红细胞平均血红蛋白浓度328 g/L (290~370 g/L);血清铁44.3 mmol/L,铁蛋白469.2 ng/mL,转铁蛋白在正常范围内;红细胞脆性实验显示在 0.50% NaCl浓度下开始溶血,在0.28% NaCl浓度下完全溶血,结果未见异常;肝肾功能、心肌酶谱等指标未见异常。骨髓细胞学显示:髓象幼红细胞罕见,其中红系比例约3%,幼红细胞罕见(早幼红未见,中幼红细胞1.5%,晚幼红细胞1.3%);粒细胞比例约为61%,淋巴细胞比例约为26%,单核细胞比例约为5%。彩色超声:左向右分流卵圆孔未闭或房间隔缺损。胸片:双肺纹理增强,未见实质性病变。
例2,女,4月龄,因“面色苍黄近并进行性加重十余天”来院就诊。入院体格检查:贫血面容,神志清楚,全身皮肤未见皮疹;双侧呼吸对称,心尖区可闻及收缩期杂音;四肢未见异常,活动无受限。实验室检查:血常规红细胞1.48×1012/L,白细胞5.61×109/L,血小板485×109/L,血红蛋白39 g/L,网织红细胞4.3×109/L,红细胞平均体积94.2 fL;血清铁41.5 mmol/L,铁蛋白491.7 ng/mL。骨髓细胞学显示:红系比例较低(<1%,早幼红细胞未见,中幼红约0.5%,晚幼红低于0.5%),有核红细胞罕见,粒细胞比例约为64%,淋巴细胞比例为28%,单核细胞约为6%,纯红细胞再生障碍性贫血(PRCA)可能性大。胸片显示双肺纹理增强。心脏彩超未见明显异常。患儿出生时无异常,父母双方均无类似疾病家族史。
为进一步明确诊断,对患儿进行DBA相关基因突变分析。经患者家属知情同意后,取患儿新鲜外周静脉血2 mL,送华大基因公司进行高通量测序检测,并利用sanger测序验证。
测序结果显示:例1的RPS19基因4号外显子上存在c.91C>T(p.P31S)杂合突变(图1)。该突变为错义突变,导致编码的RPS19蛋白在31位由脯氨酸突变为丝氨酸,患儿父母在该位点均未见突变。NCBI数据库及千人基因数据库(1000genomes)分析发现该位点不是多态性位点。RPS19基因c.91C>T(p.P31S)突变为未见报道的新发突变,利用Polyphen2软件(http://genetics.bwh.harvard.edu/pph2/)预测得分为 1.00(得分越接近1说明致病的可能性很大);SIFT(http://sift.jcvi.org/)预测的结果为0.00(越接近0,对蛋白功能影响越大)。利用不同的物种之间保守序列分析发现RPS19蛋白31位的脯氨酸十分保守,说明该位点维持蛋白正常功能具有重要的作用。
例2 的RPL5基因存在c.472_473del缺失突变,该突变会引起编码的RPL5蛋白在158位的赖氨酸处产生移码突变(p.K158fs),该位点为已知的致病性突变。患者的父母该位点未见突变(图2)。

图1 例1患儿及其父母RPS19基因检测结果
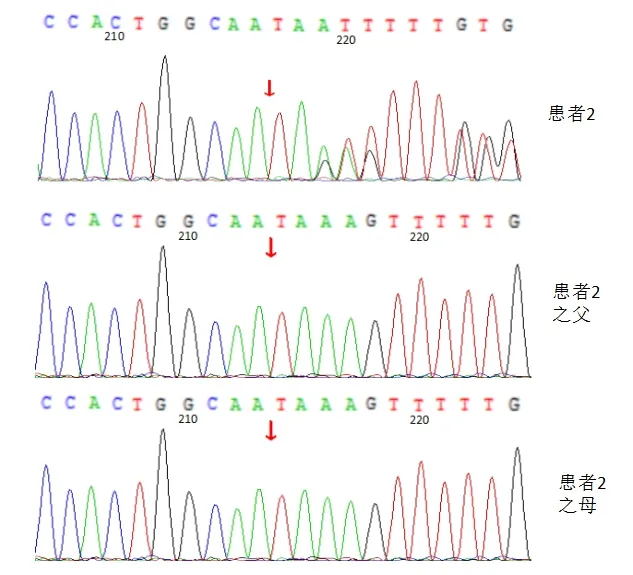
图2 例2患儿及其父母RPL5基因检测结果
2例患儿入院以后均进行相应的治疗。因患儿均不足6个月,住院期间对于贫血症状的治疗采用输注红细胞等。出院时例1的血红蛋白为86 g/L,例2为90 g/L。出院后继续口服甲泼尼龙片,4 mg/d。定期随访复查血常规,根据病情变化及时调整治疗方案。
2 讨论
DBA属于少见的遗传性骨髓衰竭性疾病中的一种,但在儿童先天性骨髓衰竭性疾病中的比例较高,仅次于范可尼贫血,现已将该病归类于核糖体疾病[7]。DBA一般在出生几个月以内起病,男女发病率约为1.1:1,大部分为散发病例,10%~25%的患者有家族史[8]。约1/3的DBA患者合并发育迟缓、尿道畸形、矮小、斜视、先天性心脏病以及拇指三指节畸形等症状。此外该类患儿易患急性非淋巴细胞白血病及非造血系统肿瘤[9,10]。DBA患者的红细胞生成障碍主要存在于红系造血集落至红系原始细胞这一阶段中[11]。
DBA的发病机制尚不完全清楚,推测可能与红系祖细胞内在性质异常、Epo信号通路异常、红系造血调控异常以及核糖体相关蛋白的功能异常有关。常规集落培养显示,DBA患者骨髓红系祖细胞(BFU-E、CFU-E)显著减少[9,12]。RPS19蛋白是第一个被发现与DBA发病相关的核糖体蛋白,该蛋白非常保守,通过与其相互作用分子发挥生物学活性,现已明确的RPS19相互作用分子包括有SI9BP、18SrRNA、Neplp、PIM-1和FGF-2[13,14]。研究发现,RPS19可能参与40S核糖体前体的组装和成熟,从而实现细胞内翻译、转运调控功能。现已经证实,单纯RPS19缺乏即可导致40S核糖体亚基的形成异常[15]。目前研究人员已经进一步建立了RPS19基因突变的斑马鱼动物模型,用于进一步探索DBA的发病机制以及新的治疗方法。RPL5基因位于1p22.1区域,其编码的蛋白属于核糖体60S的一个亚单位,且该蛋白可结合5S rRNA形成RNP,维持核糖体的正常功能[16]。
本组例1患儿,因面色苍黄近1个月入院治疗,发现其贫血严重,骨髓细胞学显示幼红细胞罕见,再根据网织红细胞等其他临床检测推测患DBA可能。国内外均有研究发现约1/3的DBA患者合并先天发育畸形等症状,例1患儿经过系统检查后发现左向右分流卵圆孔未毕或房间隔缺损。为进一步明确诊断,对患者进行DBA相关致病基因检测,发现患儿RPS19基因存在c.91C>T(p.P31S)杂合突变。根据生物信息学软件的分析预测并结合患者的临床表现,推测该位点为致病性位点,但结论需要进一步细胞水平及动物实验的验证。例2存在相似的临床表现,基因检测发现存在c.472_473del缺失杂合突变,该突变为已经证实的致病性变异[16]
研究发现,约25%的DBA患者有RPS19基因突变,2%的DBA存在RPS24基因突变[9]。另有研究表明,编码核糖体蛋白的RPS17基因、RPL5基因、RPL11基因以及RPL35a基因突变也可导致DBA的发生,此外连锁分析发现位于8p23-8p22以及1q31的未知基因可能与DBA发病相关[17]。目前高通量测序技术较为成熟,疑似患儿可进行相关致病基因检测以协助临床进行诊断。不同基因型的异常可能导致不同的临床表型。研究发现,RPL5与RPL11基因突变与DBA畸形发生相关[18];核糖体蛋白基因突变阳性的DBA患儿畸形发生率>40%,明显高于无突变患儿,其中携带RPL5与RPL11基因突变者易伴有指/趾畸形,而携带RPS19基因突变者易产生尿道畸形[6]。本组2例患儿未见相似畸形发生。
DBA的治疗手段主要以糖皮质激素、红细胞输注以及造血干细胞移植为主,其中糖皮质激素为DBA的一线治疗药物[19]。国内对50例接受泼尼松治疗的DBA患儿分析显示,其治疗反应率为64.0%,多数患儿治疗1个月以内见效,15例患儿获得缓解,其中5例停药,10例口服泼尼松维持治疗[6]。DBA患儿对糖皮质激素反应的差异可能与机体内糖皮质激素受体的多态性相关[20],今后可对此进行遗传学差异分析。
综上所述,DBA患儿起病年龄较早,多在1岁以内,临床表现为巨幼红细胞性贫血、网织红细胞数减少、红系细胞明显缺乏等,核糖体相关蛋白基因突变为导致DBA的常见病因。基因c.91C>T(p.P31S)位点突变未见报道,依据患儿的相关临床表现及针对编码核糖体蛋白的基因突变筛查有利于DBA的诊断及尽早治疗。
[1]Horos R,von Lindern M.Molecular mechanisms of pathology and treatment in Diamond Blackfan Anaemia [J].Br J Haematol,2012,159(5): 514-527.
[2]Narla A,Vlachos A,Nathan DG.Diamond Blackfan anemia treatment: past,present,and future [J].Semin Hematol,2011,48(2): 117-123.
[3]Sakaguchi H,Nakanishi K,Kojima S.Inherited bone marrow failure syndrome in 2012 [J].Int J Hematol,2013,97(1): 20-29.
[4]Draptchinskaia N,Gustavsson P,Andersson B,et al.The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia [J].Nat Genet,1999,21(2): 169-175.
[5]Vlachos A,Dahl N,Dianzani I,et al.Clinical utility gene card for: Diamond-Blackfan anemia-update 2013 [J].Eur J Hum Genet,2013,21(10): 1-4.
[6]刘天峰,万杨,陈玉梅,等.Diamond-Blackfan贫血患儿的临床分析 [J].国际输血及血液学杂志,2014,37(5):406-411.
[7]Quarello P,Garelli E,Carando A,et al.Ribosomal RNA analysis in the diagnosis of Diamond-Blackfan anaemia [J].Br J Haematol,2016,172(5): 782-785.
[8]陈玉梅,阮敏,邹尧,等.21例先天性纯红细胞再生障碍性贫血核糖体蛋白基因突变分析[J].中国实验血液学杂志,2012,20(6): 1414-1418.
[9]郑杰,吴润晖.Diamond-Blackfan贫血临床和分子生物学研究进展[J].中国实用儿科杂志,2009,24(2):148-150.
[10]Vlachos A,Ball S,Dahl N,et al.Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference [J].Br J Haematol,2008,142(6): 859-876.
[11]Wan Y,Zhang Q,Zhang Z,et al.Transcriptome analysis reveals a ribosome constituents disorder involved in the RPL5 downregulated zebra fi sh model of Diamond-Blackfan anemia[J].BMC Med Genomics,2016,9: 13.
[12]Farrar JE,Quarello P,Fisher R,et al.Exploiting pre-rRNA processing in Diamond-Blackfan anemia gene discovery and diagnosis [J].Am J Hematol,2014,89(10): 985-891.
[13]Solomon J,Kamalammal R,Menezes GA,et al.A case of diamond blackfan anemia (DBA) with mutation in ribosomal protein S19 [J].J Clin Diagn Res,2014,8(1): 179-180.
[14]Orrù S,Aspesi A,Armiraglio M,et al.Analysis of the ribosomal protein S19 interactome [J].Mol Cell Proteomics,2007,6(3): 382-393.
[15]Choesmel V,Bacqueville D,Rouquette J,et al.Impaired ribosome biogenesis in Diamond-Blackfan anemia [J].Blood,2007,109(3): 1275-1283.
[16]Quarello P,Garelli E,Carando A,et al.Diamond-Blackfan anemia: genotype-phenotype correlations in Italian patients withRPL5andRPL11mutations [J].Haematologica,2010,95(2): 206-213.
[17]Chiabrando D,Tolosano E.Diamond Blackfan anemia at the crossroad between ribosome biogenesis and heme metabolism[J].Adv Hematol,2010,2010: 1-8.
[18]Gazda HT,Sheen MR,Vlachos A,et al.Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients [J].Am J Hum Genet,2008,83(6): 769-780.
[19]Vlachos A,Blanc L,Lipton JM.Diamond Blackfan anemia: a model for the translational approach to understanding human disease [J].Expert Rev Hematol,2014,7(3): 359-372.
[20]Varricchio L,Godbold J,Scott SA,et al.Increased frequency of the glucocorticoid receptor A3669G (rs6198) polymorphism in patients with Diamond-Blackfan anemia [J].Blood,2011,118(2): 473-474.
2017-02-15)
(本文编辑:梁 华)
Clinical features of Diamond-Blackfan anemia and gene testing
SHI Yumei,MA Jie(Department of Paediatrics,Shanxian Hygeia Hospital,Shanxian 274300,Shandong,China)
ObjectiveTo investigate the clinical and genetic features of Diamond-Blackfan anemia (DBA).MethodThe clinical manifestations and genetic tests of 2 cases with DBA were retrospectively analyzed,and the related literatures were reviewed.ResultsTwo female patient (3-4 month old)with progressive ochriasis nearly a month was included.Fever,seizure,vomit and abnormal change in urine and stool routine test were not shown.Blood routine test: the number of RBC in the two patients was decreased(1.24 ×1012/L and 1.48×1012/L),HGB (46 g/L and 39 g/L),and the number of RTC was also decreased(4.1×109/L and 4.3×109/L),RCV was normal (108.4 fl).Serum iron determination: Fe (44.3 mmol/L and 41.5 mmol/L) and ferritin (469.2 mmol/L and 491.7 ng/mL) were increased,transferrin was in the normal range.Erythrocyte fragility test resulted normal.Bone marrow examination found rarely erythroblasts.A novel heterozygous mutation inRPS19gene,c.91C>T(p.P31S),was found by genetic testing on patient 1.And we found a heterozygous mutation inRPL5gene (c.472_473del) in patient 2.Conclusion The majority of onset age of childhood DBA was within a few months with a erythroid de fi ciency.AndRPS19gene mutation is a common cause of this disease.The mutation of c.91C>T (p.P31S) has not been reported.
Diamond-Blackfan anemia; clinical manifestations; genetic testing
10.3969/j.issn.1000-3606.2017.11.012
