33例中国型Gγ(Aγδβ)0地中海贫血病例的临床分析
2017-11-25陈剑虹钟泽艳官志扬贺海林钟国兴杨坤祥
陈剑虹 钟泽艳 官志扬 贺海林 钟国兴 杨坤祥
33例中国型Gγ(Aγδβ)0地中海贫血病例的临床分析
陈剑虹★钟泽艳 官志扬 贺海林 钟国兴 杨坤祥
目的 探讨惠州地区人群中国型Gγ(Aγδβ)0地中海贫血的检出率及临床特征,分析其意义及完善地贫防控内容。 方法 2015年1月至2016年12月,对参加惠州市孕前和孕期优生健康检查育龄夫妇的24137例血液样本行血常规及血红蛋白电泳检测,并应用液相芯片、导流杂交技术对检出结果分析为可疑地中海贫血样本及产前363例羊水样本进行地中海贫血基因检测,对于疑为β地中海贫血但未检出者加用Gap⁃PCR技术检测中国型Gγ(Aγδβ)0地中海贫血缺失型基因。 结果 在290例加测样本中检出中国型Gγ(Aγδβ)0杂合子29例,中国型Gγ(Aγδβ)0地中海贫血复合α地中海贫血4例,总检出率为0.14%。血常规结果显示血红蛋白正常为26例,轻度下降为7例;红细胞平均体积(mean corpuscular volume,MCV)及红细胞平均血红蛋白含量(mean corpuscular hemoglobin,MCH)均降低;血红蛋白电泳结果显示HbA2正常或轻度降低,HbF升高为(14.56±5.43)%。检出4例为夫妻同型β地贫携带者,行产前诊断,一例胎儿基因型为 β⁃28杂合突变复合中国型Gγ(Aγδβ)0杂合缺失,其中国型Gγ(Aγδβ)0基因遗传自母方,β⁃28基因遗传自父方,终止妊娠。 结论 惠州地区人群的中国型Gγ(Aγδβ)0携带率高,中国型Gγ(Aγδβ)0携带者的临床特征为MCV、MCH降低的基础上HbF值明显升高;中国型Gγ(Aγδβ)0属缺失型β地中海贫血类型,当其有可能与β地中海贫血复合存在时,应进行产前诊断,防止中重型地贫患儿出生。
地中海贫血; 中国型Gγ(Aγδβ)0; 基因检测; 产前诊断
地中海贫血(地贫),又称珠蛋白生成障碍性贫血,是由于珠蛋白基因缺陷导致珠蛋白链合成障碍而引起的一种遗传性溶血性贫血。地贫具有显著的地域分布性,为我国南方最常见和危害最大的遗传病之一,主要分为α、β、δ、γ和δβ地贫。δβ地贫是一组因基因发育阶段特异性表达失控导致胎儿出生后HbF持续增高的地贫,其大片段缺失型常见型别有中国型、广州型、东南亚型、云南型及中国台湾型,其中我国最常见的δβ⁃地中海贫血为中国型Gγ(Aγδβ)0地贫[1⁃3]。广东省大规模人群地贫携带率的基线调查中显示,惠州市的地贫携带率(17.5%)明显高于平均水平(16.83%)[4]。因此,地贫防控对于提高惠州市人口素质具有重要意义。本研究在国家卫生和计生委实施“国家免费孕前优生健康检查项目”的背景下,在惠州市政府“民生工程”的支持下,以惠州市育龄夫妇人群为对象,对孕前和孕期地贫初筛异常夫妻进行免费血红蛋白电泳分析及常规地贫基因检测,并为同型地贫夫妇提供遗传咨询指导、追踪随访等服务。本课题对惠州地区育龄夫妇中国型Gγ(Aγδβ)0地贫基因携带率及其临床特征等流行病学资料进行分析,补充本地区人群地贫防控内容,更有效地指导预防中重型地贫患儿的出生。
1 对象与方法
1.1 研究对象
于2015年1月至2016年12月,根据知情同意的原则,采集参加惠州市孕前和孕期优生健康检查育龄夫妇的样本共24137例,年龄范围为20~60岁,平均年龄为26岁,近3个月无输血史。每人均抽取外周静脉血2 mL置于EDTA、ACD抗凝管,用于血常规、血红蛋白电泳分析,和提取外周血基因组DNA用于地贫基因检测。同时,本中心对可能会分娩中重型地贫患儿的孕妇行羊膜腔穿刺术。采集胎儿羊水样本2管各约10 mL,一管直接法检测(培养前),另一管按照本中心建立的常规方法进行细胞培养后检测。
1.2 研究方法
1.2.1 血液学筛查
采用深圳迈瑞生物医疗电子股份有限公司的BC⁃5380全自动五分类血液细胞分析仪检测红细胞参数,包括血红蛋白(hemoglobin,HGB)、红细胞平均体积(mean corpuscular volume,MCV)及红细胞平均血红蛋白含量(mean corpuscular hemoglobin,MCH)等。初筛阳性标准为:MCV<82.00 fL或(和)MCH<27.00 pg[1,4,17]。采用法国 SEBIA 公司的Capillarys 2 flex piercing全自动毛细管电泳仪分析血红蛋白A2(hemoglobin A2,HbA2)、血红蛋白F(hemoglobin F,HbF)和异常血红蛋白条带,分别以HbA2<2.50%或(和)出现异常血红蛋白带,例如血红蛋白H(hemoglobin H,HbH)、血红蛋白CS(hemoglobin constant spring,HbCS)等,判定为α地贫表型阳性,HbA2>3.50%或(和)HbF>2.50%判定为 β 地贫表型阳性[1,4,17]。
1.2.2 基因型分析
对血液学筛查为阳性的样本以及产前诊断的胎儿羊水样本,按全血基因组DNA提取试剂盒说明书用Lab⁃Aid 820核酸提取仪(厦门百维信生物公司)提取基因组DNA。获得的DNA用液相芯片技术[地中海贫血(α/β型)基因检测试剂盒,中山大学达安基因股份有限公司]和导流杂交技术(α⁃和β⁃地贫基因检测试剂盒,潮州凯普生物化学有限公司)进行基因诊断,检测范围为中国人常见的3种α缺失型地贫(⁃⁃SEA/、⁃α4.2/、⁃α3.7/),3种α非缺失型地贫(αWSα/、αCSα/、αQSα/),17 种 β 地 贫(CD41 ⁃42M、CD17M、CD43M、CD71⁃72M、IVS⁃I⁃1M、IVS⁃I⁃5M、IVS⁃II⁃654M、⁃28M、⁃29M、⁃30M、⁃32M、CapM、IntM、CD14⁃15M、CD27⁃28M、βEM、CD31M)。按照操作说明书进行操作和结果判断。对疑为β地贫而未检出常见突变基因的样本,进一步采用裂隙聚合酶链反应(gap polymerase chain reaction,Gap⁃PCR)技术检测中国型Gγ(Aγδβ)0缺失型,目的片段长度约300 bp。
2 结果
2.1 常规基因型结果
在24137例血液样本中,根据血液学筛查阳性样本的纳入标准,判定为β地贫表型阳性而未检出常规基因型的样本有290例,所占比例为1.20%。363例产前羊水样本检出1例异常遗传规律的胎儿基因型为 β⁃28/β⁃28、⁃⁃SEA/αα(图 1A),其父亲基因型为 β⁃28/βN、⁃⁃SEA/αα(图 1B),母亲基因型为βN/βN、αα/αα(图1C),见图1和表1。
2.2 加测基因型结果
290 例加测样本中,检出中国型Gγ(Aγδβ)0地贫 33例,检出率为 0.14%。其中中国型Gγ(Aγδβ)0地贫杂合子29例,复合α地贫共4例,分别为2例⁃α3.7/αα、1 例⁃⁃SEA/αα、1 例 αQSα/αα。33 例中国型Gγ(Aγδβ)0地贫杂合子均表现为小细胞低色素,MCV 为(72.47±5.00)fL,MCH 为(24.01±2.45)pg;血红蛋白正常为26例,轻度下降为7例;HbA2正常或轻度下降,HbF明显升高为(14.56±5.43)%,见表2。检出4例的配偶同为β⁃地贫携带者(样本1~4号),属夫妻同型地贫携带者,进行产前诊断。其中检出一例胎儿常规基因型结果显示遗传规律异常,提示为中重型地贫患儿,见图2。

图1 异常遗传规律家系导流杂交结果Figure 1 The hybri Max results of abnormal genetic rule family
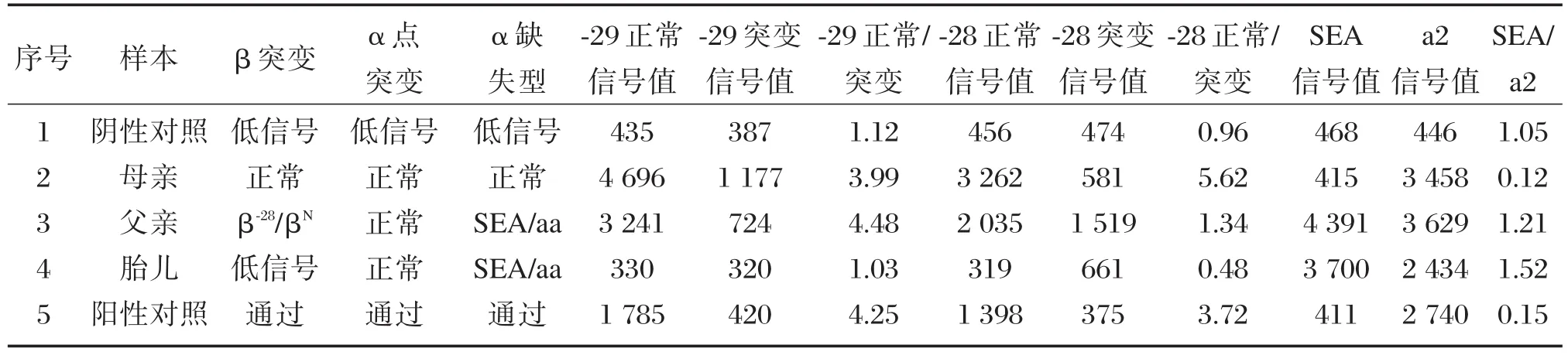
表1 异常遗传规律家系液相芯片结果Table 1 The liquid chip results of abnormal genetic rule family
3 讨论
中国型Gγ(Aγδβ)0地贫最初于20世纪70年代由Mann等[5]首次报道,其缺失长度约为100 kb,累及β珠蛋白基因、Aγ、ψβ、δ和β珠蛋白基因下游较远的具有调控功能的 DNA 序列[6⁃7]。β 珠蛋白基因簇中含有2个表达γ⁃珠蛋白肽链的γ⁃基因(Gγ和Aγ),因此这种缺失致使其杂合子除一般β地贫的小细胞低色素性表型外,还伴有Aγ、Gγ珠蛋白基因的高表达以及HbF异常升高等特征性表现。HbF是胎儿期表达的血红蛋白,胎儿出生后基因表达关闭,HbA(α2β2)成为红细胞中主要的血红蛋白,因此 HbF含量比率少于1%[1⁃3]。与 β 地贫杂合子或双重杂合子一样,此类缺失突变和β地贫点突变的复合型亦可导致中重型地贫[3,8]。预防中重型地贫患儿出生最有效的方法是产前筛查出地贫基因携带者,对夫妻同型这类高风险家庭进行产前诊断,从而达到优生优育的目标[9⁃10]。
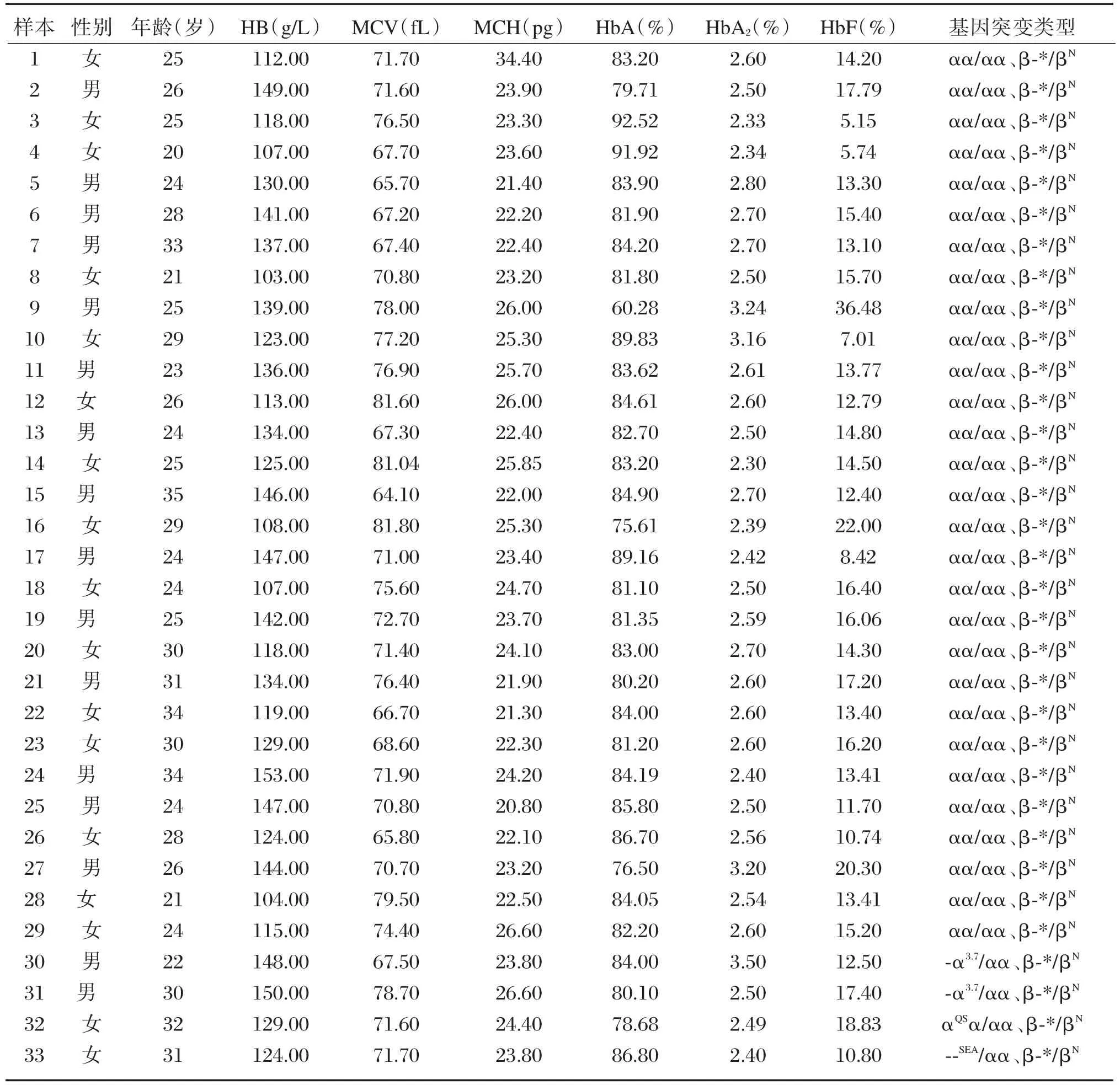
表2 33例中国型Gγ(Aγδβ)0地中海贫血标本的血液学参数及基因型Table 2 The hematological parameters and genotypes of 33 ChineseGγ(Aγδβ)0⁃thalassemia specimens
本研究中,惠州地区24137例样本中检出33例中国型Gγ(Aγδβ)0地贫缺失杂合子患者,携带率高达0.14%,比已报道的广西地区携带率0.07%高[11]。中国型Gγ(Aγδβ)0地贫携带者的红细胞参数结果显示患者表现为无贫血或轻度贫血,血红蛋白值为 103~153 g/L,但 MCV<82.00 fL、MCH<27.00 pg均显示小细胞低色素贫血的特征,提示地贫的可能,这与文献报道的结果相类似[3,11⁃13]。在血红蛋白电泳结果中,HbA2在正常范围(2.5%~3.5%)或轻微降低,而HbF则明显升高(5.15%~36.48%),与王继成等[3](10.6%~20.6%)、韦媛等[11](10.8%~22.3%)及覃丽波[12](10%~18%)报道的情况相比,血液学参数范围更大。分析原因,可能是本研究病例较多导致的,本中心的标本除了在本院就诊的病人,还包括采集于下属多个县、区医院,血液学的检查没有统一的质控,所用的仪器也非统一。此外,也可能由于惠州地区有少数民族,存在个体差异性大,其中HbF较高的样本来自少数民族较多的龙门县区。目前关于惠州地区少数民族的研究数据较少,以后可有针对性地收集少数民族样本,总结归纳该群体的基因分布情况和表型特征。虽然中国型Gγ(Aγδβ)0地贫缺失杂合子患者的临床症状较轻,但当复合β地贫时,患者会出现中度至重度贫血,临床表型类似于中重型β 地贫[3,11⁃14]。目前国内商业化的地贫基因试剂盒仅检测17种最常见的β地贫,不包括中国型Gγ(Aγδβ)0在内的缺失型β地贫。因此,进行血液学分析时,若红细胞指标提示为小细胞低色素性贫血,且行常规地贫基因检测为阴性结果,同时HbF有一定程度的升高,特别是当HbF>5%时,应考虑缺失型β地贫的可能[13⁃16]。同理,当一个家系中出现重型β地贫患者,但用常规方法只能找到一个基因突变时,或夫妻双方基因型与重型地贫患儿基因结果不符合时,应考虑缺失突变的可能,需用合适的方法验证,并行产前诊断。所以在临床遗传咨询中必须警惕夫妻一方已确诊是β地贫基因携带者,当配偶的地贫筛查中出现HbF明显增高,要采用其他方法进一步排除罕见型地贫。总之,应注意与重视罕见型地贫的筛查与诊断,以避免漏诊或误诊,从而更好地指导临床诊断。
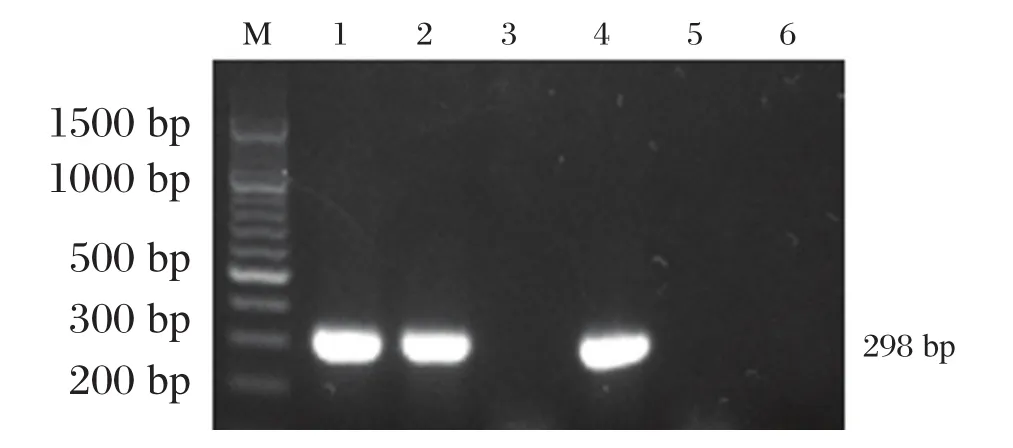
图2 异常遗传规律家系中国型Gγ(Aγδβ)0地中海贫血结果Table 2 The ChineseGγ(Aγδβ)0⁃thalassemia results of abnormal genetic rule family
本研究检测出的患者为中国型Gγ(Aγδβ)0地贫且其配偶为β地贫者4例,均进行产前诊断,分别检测出2例正常胎儿、1例β地贫携带者及1例中国型Gγ(Aγδβ)0地贫复合 β 地贫双重杂合子胎儿。其中复合β地贫的胎儿常规基因型结果显示遗传规律异常,提示为中重型地贫患儿。经遗传咨询后,该例患者选择终止妊娠。比较液相芯片和导流杂交 2 种技术检测中国型Gγ(Aγδβ)0地贫复合β地贫双重杂合子的结果图发现,图1的杂交膜条A中的⁃28位点无正常对照,显示为该位点的纯合突变;表1的胎儿样本结果显示⁃28和⁃29突变点均为低信号值。分析原因为导流杂交技术设置的⁃28和⁃29位点共用⁃28正常对照点,而液相芯片技术⁃28和⁃292个突变相邻近,因存在大片段缺失导致分析软件无法正常判读。因此,目前国内商业化的试剂盒虽无法检出中国型Gγ(Aγδβ)0地贫,但若与β地贫复合存在时,异常结果可提示需对样本进行加测确诊。从本研究检出 4 例中国型Gγ(Aγδβ)0地贫复合 α 地贫双重杂合子中,发现单独的中国型Gγ(Aγδβ)0地贫缺失杂合子和其复合α地贫基因突变时,血液学表型特征无明显差异性。
惠州地区是地贫高发区[4,17],特别是针对于罕见β地贫的正确诊断,可为遗传咨询和产前诊断提供更好的依据。本研究数据为该地区地贫的流行病学调查提供了新的佐证,完善了地贫防控内容,从而能更有效地避免中重型地贫儿的出生,提高出生人口素质,为家庭、社会减轻负担,同时也提高人们的生活质量。
[1] 徐湘民.见于中国人的HPFH和δβ⁃地中海贫血的分子基础[J].中华医学遗传学杂志,1998,15(5):315⁃317.
[2] Carrocini GC,Ondei LS,Zamaro PJ,et al.Evalua⁃tion of HPFH and δβ⁃thalasemia mutations in a Brazil⁃ian group with high HbF levels[J].Genet Mol Res,2011,10(4):3213⁃3221.
[3] 王继成,秦丹卿,骆明勇,等. 中国型Gγ+(Aγδβ)0地中海贫血的基因诊断和临床特征分析[J].分子诊断与治疗杂志,2016,8(4):227⁃230.
[4] Yin A,Li B,Luo M,et al.The Prevalence and Mo⁃lecular Spectrum of a⁃and b⁃Globin Gene Mutations in 14,332 Families of Guangdong Province,China[J].PLoS One,2014,9(2):e89855.
[5] Mann JR,MacNeish AS,Bannister D,et al.Delta⁃be⁃ta⁃thalassemia in a chinese family[J].Br J Haematol,1972,23(4):393⁃402.
[6] Fornari TA,Lanaro C,Albuquerque DM,et al.Mod⁃ulation of fetal hemoglobin in hereditary persistence of fetal hemoglobin deletion type⁃2,compared to Sicilian δβ⁃thalassemia,by BCL11A and SOX6⁃targeting mi⁃croRNAs[J].Exp Biol Med(Maywood),2017,242(3):267⁃274.
[7] Dehury S,Purohit P,Meher S,et al.Compound het⁃erozygous state of β⁃thalassemia with IVS1⁃5(G→C)mutation and Indian deletion ⁃inversionGγ(Aγδβ)0⁃thalassemia in eastern India[J].Rev Bras Hematol He⁃moter,2015,37(3):202⁃206.
[8] 朱恒莹,陈萍.非缺失型遗传性持续性胎儿血红蛋白综合征复合β地中海贫血的研究进展[J].实用医学杂志,2016,32(19):3268⁃3270.
[9] 江帆,唐盈,陈桂兰,等.广州地区孕前人群地中海贫血基因型分布状况及干预模式探讨[J].中国优生与遗传杂志,2016(8):7⁃10.
[10] 谭建强,潘莉珍,陆碧玉,等.柳州市中重度地中海贫血患儿出生原因调查[J].中国妇幼保健,2017,32(1):135⁃138.
[11]韦媛,林丽,陈碧艳,等.广西地区中国型Gγ(Aγδβ)0⁃地中海贫血的发病情况及表型特征[J].山东医药,2016,56(37):58⁃60.
[12] 覃丽波,陈萍,陈文强,等. 广西地区Gγ(Aγδβ)0⁃珠蛋白生成障碍性贫血的研究[J].实用儿科临床杂志,2012,27(15):1151⁃1153.
[13] 杜丽,秦丹卿,王继成,等.5个家系δβ地中海贫血的家系分析及产前诊断[J].分子诊断与治疗杂志,2015,7(1):27⁃32.
[14] Mondal S K,Mandal S.Prevalence of thalassemia and hemoglobinopathy in eastern India:A 10⁃year high⁃performance liquid chromatography study of 119,336 cases[J].Asian J Transfus Sci,2016,10(1):105⁃110.
[15] 林开颜,张晋,殷立,等.全自动毛细管血红蛋白电泳对育龄夫妇地中海贫血筛查的诊断价值[J].中国医药指南,2016,14(34):28⁃29.
[16] 郑琳,黄海龙,范向群,等.血细胞和血红蛋白分析在地中海贫血中的应用价值[J].中国妇幼保健,2016,31(3):547⁃549.
[17] 钟泽艳,陈剑虹,贺海林,等.广东省惠州地区基于医院水平的地中海贫血基因突变谱分析[J].中国计划生育学杂志,2015,23(6):371⁃375.
Clinical analysis of 33 cases of deletional ChineseGγ (Aγδβ)0⁃thalassemia in Huizhou
CHEN Jianhong★,ZHONG Zeyan,GUAN Zhiyang,HE Hailin,ZHONG Guoxing,YANG Kunxiang
(Prenatal Diagnosis Center of Huizhou First Women and Children's Hospital,Huizhou,Guangdong,China,516007)
Objective To Explore the gene frequency and clinical features of ChineseGγ(Aγδβ)0⁃thalassemia in Huizhou,to analyze its significance and improve the prevention and control of thalassemia.Methods From January 2015 to December 2016,a total of 24137 blood samples were collected from couples of childbearing age who participated in the pre⁃pregnant and prenatal healthy births in Huizhou city and analyzed by using blood routine test and hemoglobin electrophoresis as suspicious thalassemia screening.363 amniotic fluid samples were detected by the liquid phase chip and diversion hybridization technique for thalassemia simultaneously.Further,the Gap⁃PCR technique was used to detect the gene of deletional ChineseGγ(Aγδβ)0⁃thalassemia for undetected patients with suspected β⁃thalassemia. Results 29 cases of ChineseGγ(Aγδβ)0heterozygotes and 4 cases of ChineseGγ(Aγδβ)0⁃thalassemia accompanied by α⁃thalassemia were identified in 290 samples with the total detection rate of 0.14%.The blood routine test results showed that there were 26 cases of normal hemoglobin,and 7 cases of slight decrease.The mean corpuscular volume(MCV)andthemeancorpuscularhemoglobin(MCH)weredecreased.Theresultsofhemoglobin electrophoresis showed that HbA2was normal or slight decrease,with the HbF increased to(14.56±5.43)%.4 cases of couples with the same genotypes of thalassemia were taken by prenatal diagnosis.One Case fetal abnormal genotype was ChineseGγ(Aγδβ)0⁃thalassemia accompanied by β⁃28.The ChineseGγ(Aγδβ)0gene inherited from mother and the β⁃28gene inherited from father and this pregnancy was terminated.Conclusion The incidence of Chinese typeGγ(Aγδβ)0⁃thalassemia in Huizhou area is high.The clinical features of Chinese typeGγ(Aγδβ)0⁃thalassemia carriers have both MCV and MCH decrease and HbF increase.The Chinese typeGγ(Aγδβ)0belongs to the deletional β ⁃thalassemia.The prenatal diagnosis should be performed to prevent the birth of children with moderate or severe thalassemia when the ChineseGγ(Aγδβ)0⁃thalassemia is accompanied by α⁃thalassemia.
Thalassemia;ChineseGγ(Aγδβ)0;Gene detection;Prenatal diagnosis
广东省惠州市第一妇幼保健院产前诊断中心,广东,惠州516007
★通讯作者:陈剑虹,E⁃mail:1296275774@qq.com