原发性胆汁性胆管炎的动物模型
2017-11-22刘成海
郝 娟, 刘成海,2,3
(1 上海中医药大学附属曙光医院 肝病研究所, 上海 201203; 2 上海市中医临床重点实验室, 上海 201203;3 上海高校中医内科学 E-研究院, 上海 201203)
原发性胆汁性胆管炎的动物模型
郝 娟1, 刘成海1,2,3
(1 上海中医药大学附属曙光医院 肝病研究所, 上海 201203; 2 上海市中医临床重点实验室, 上海 201203;3 上海高校中医内科学 E-研究院, 上海 201203)
理想的原发性胆汁性胆管炎(PBC)动物模型对于研究疾病的病理生理机制与药物研发等均有重要意义。近年来可反映血清抗线粒体抗体阳性与胆管免疫病理损伤等PBC特点的动物模型取得了较大进展。目前PBC动物模型的制备方式多样,包括化学或生物染毒诱导,以及基因敲除后自发形成。但上述模型尚无法完全模拟人类PBC,其血清学、免疫学、组织病理等方面也各有特点,提示PBC基因特异与环境改变的病理机制的复杂性,对认识免疫耐受性被打破和胆管上皮细胞受到特异性攻击等PBC早期事件的机制具有重要意义。
胆管炎, 胆汁性; 模型, 动物
原发性胆汁性胆管炎(PBC)是器官特异性自身免疫性疾病,其特征在于约95%的PBC患者血清高滴度抗线粒体抗体(AMA)和进行性门静脉周围淋巴细胞介导的胆管上皮细胞免疫性破坏[1]。PBC以中老年女性高发,但病因机制不清。目前,PBC诊断仍是一种排他性诊断方式,即具备以下3条标准中的2条:(1)存在胆汁淤积的生化学表现,如ALP水平升高;(2)血清抗线粒体抗体(AMA)或AMA-M2阳性;(3)肝组织病理显示非化脓性胆管炎与小叶间胆管损伤[2],这也透露出PBC的病理机制研究存在较多疑点和难点。动物模型是阐述人类疾病病因病理机制、开发治疗药物的重要工具。目前,几种PBC的动物模型通过基因修饰或化学生物免疫诱导2种方式模拟PBC的血清学、免疫学或病理学特征。这些动物模型有助于剖析遗传、环境因素在PBC发病中的作用,促进对PBC发病过程的理解,现分述如下。
1 基因修饰类模型
1.1 dnTGFβ RⅡ小鼠模型 TGFβ Ⅱ型受体显性失活(dominant-negative transforming growth factor-β receptor type Ⅱ,dnTGFβ RⅡ)转基因小鼠模型最初由R. A. Flavell开发,用于研究该受体在T淋巴细胞功能中的作用[3]。在CD4启动子的控制下,dnTGFβ RⅡ转基因小鼠过表达TGFβ受体Ⅱ型显性抑制基因。
该模型的表型特征包括:(1)针对相同的线粒体自身抗原丙酮酸脱氢酶的E2亚基(the E2 subunits of pyruvate dehydrogenase,PDC-E2)、支链2-氧代酸脱氢酶(branched chain 2-oxo acid dehydrogenase,BCOADC-E2),2-氧代-戊二酸脱氢酶(2-oxo-glutarate dehydrogenase,OGDC-E2)自发产生AMA,阳性率分别为100%、68%和95%;(2)含有AMA的血清能够体外特异性抑制PDC-E2酶活性;(3)肝实质和门静脉区有特异性巨噬细胞和淋巴细胞浸润,主要包括CD4+、CD8+、B淋巴细胞和自然杀伤细胞(NK细胞);(4)胆管上皮细胞破坏严重,有些甚至不能确定完整的胆管结构;(5)相似的血清细胞因子谱:血清中IFNγ、TNFα、IL-6和IL-12 p40水平均显著增加[4](表1)。
dnTGFβ RⅡ小鼠模型不同于人类PBC的特点有:(1)发病无性别差异;(2)血清ALP无法测得;(3)肝内无嗜酸性粒细胞浸润,肉芽肿形成也不明显;(4)肝内CD3+淋巴细胞总数和比例均明显增高;CD4+淋巴细胞总数有所增加,但比例增加不明显;CD8+淋巴细胞总数和比例均有显著升高;(5)免疫球蛋白以IgA升高为主[4]。
然而,同型对照dnTGFβRⅡRag1-/-小鼠血清中不能检测到AMA,肝组织学显示正常,无肝细胞或胆管特异性病理损伤证据[4]。可见,dnTGFβRⅡ是引发该模型病理机制的关键因素,TGFβRⅡ通路对PBC发病机制具有重要的提示作用。
TGFβ具有调节细胞增殖、分化和迁移等多种功能,在调节炎症、伤口修复、免疫稳态和耐受性方面起着重要作用[5]。TGFβ缺乏可导致多种自身免疫性疾病,如自身免疫性胆管炎和结肠炎等。TGFβ受体Ⅱ对TGFβ信号转导非常重要,可调节淋巴细胞的活化。显性负性TGFβ受体是由CD4+和CD8+T淋巴细胞表达的,可引起TGFβ信号传导大大降低,导致T淋巴细胞固有细胞介导的自身免疫反应[6]。为了进一步明确T淋巴细胞在PBC发病中的作用,将dnTGFβ RⅡ来源的CD8+T淋巴细胞转移到Rag1-/-小鼠体内,结果导致了肝脏特异的自身免疫性胆管炎;而将CD4+T淋巴细胞转移到Rag1-/-小鼠体内形成了结肠炎,提示CD8+T淋巴细胞是dnTGFβ RⅡ小鼠发生胆管损伤的主要贡献者[7]。因此,缺陷的TGFβ RⅡ信号传导和以胆管细胞为靶点的抗原特异性克隆CD8+T淋巴细胞是诱导自身免疫性胆管炎所必需的[8]。
1.2 NOD.c3c4小鼠模型 非肥胖型糖尿病(non obesity diabetic,NOD)小鼠原本用于研究自身免疫性1型糖尿病。NOD.c3c4同源小鼠是在NOD小鼠背景下,将B6/B10来源的胰岛素依赖性糖尿病(insulin-dependent diabetes,Idd)抗性等位基因分别插入到NOD小鼠的3、4号染色体(c3.c4)上,以阻止糖尿病的发生。然而,实验观察到仅染色体3上具有B6抗性等位基因的NOD小鼠表现出肝内有淋巴细胞浸润,但无自身抗体形成;仅染色体4上具有B10抗性等位基因的NOD小鼠肝内无淋巴细胞浸润,但有自身抗体形成[9]。可见,虽然保护性c3/c4基因座的组合可以有效预防糖尿病,但B6和B10抗性等位基因与NOD基因组发生交互作用后,可导致一种新型的遗传控制的自身免疫性肝病。
NOD.c3c4同源小鼠的表型特征:(1)血清ALT和AST升高;(2)因胆道疾病引起肝肿大,甚至肝衰竭;(3)胆道梗阻症状逐渐加重,导致腹水形成;(4)50%~60%的NOD.c3c4小鼠在9~10周时自发产生针对PDC-E2的自身抗体AMA;80%~90%的NOD.c3c4小鼠在20周时出现血清ANA阳性,最显著的是抗Sm抗体;(5)肝内胆管囊肿进行性形成,伴有囊肿壁周围淋巴细胞浸润,以CD3+、CD4+、CD8+T淋巴细胞为主;(6)上皮样肉芽肿形成;(7)胰腺出现轻度炎症和唾液腺内有淋巴细胞浸润;(8)自身免疫性胆管炎可以通过免疫细胞如脾细胞或CD4+T淋巴细胞转移至NOD.c3c4免疫缺陷小鼠[9-10](表1)。
该模型与人类PBC的不同点表现在NOD.c3c4小鼠发病无性别差异,ALP无法测得,Ig以IgA升高为主,AMA的阳性率约为60%,具有比人类PBC更加广泛的胆管增生改变和自身免疫攻击部位,NOD.c3c4小鼠以胆总管和肝内胆管为靶标,而PBC主要攻击肝内中小胆管[11]。
人类PBC的组织学特征为早期胆管周围肉芽肿形成、门静脉周围嗜酸粒细胞浸润和肝内小胆管破坏,称为非化脓性破坏性胆管炎。Terasaki等[12]发现PBC早期的嗜酸粒细胞增多与胆管周围淋巴细胞浸润、肉芽肿形成和胆管损伤呈正相关。NOD.c3c4小鼠组织学以肝内胆管多囊性病变为特点,与人类PBC相同的变化是二者均为非化脓性破坏性胆管炎,另可见到损伤胆管周围嗜酸性粒细胞浸润和上皮样肉芽肿病变[10]。
1.3 AE2a,b-/-小鼠模型 Cl-/HCO3-阴离子交换器2(Cl-/HCO3-anion exchanger 2,AE2)是一种广泛表达的膜溶质载体,为电中性的Na+依赖性交换器,具有调节细胞内pH稳态、Cl-浓度和细胞体积等作用[13-14]。在胆管细胞中,碳酸氢盐分泌由AE2介导。在极化上皮细胞中,AE2有助于上皮细胞分泌和重吸收酸碱等同物和Cl-;细胞内或细胞外H+可紧急独立抑制AE2,该调节需要AE2 N末端胞质结构域的最高度保守序列的完整性[15-16]。
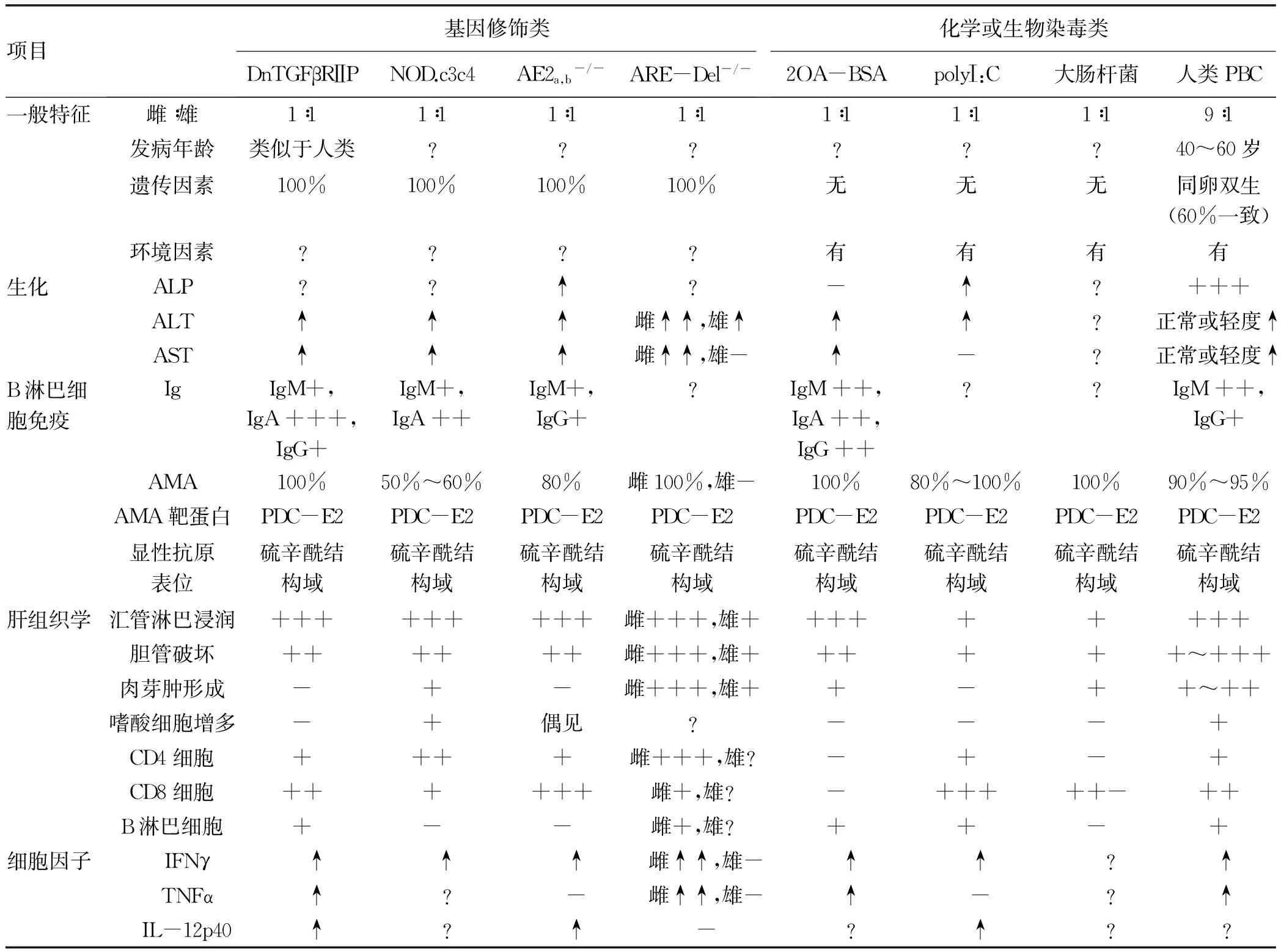
表1 人类PBC与PBC动物模型的特点比较
AE2a,b-/-小鼠的表型特征:(1)脾脏肿大;(2)血清AMA阳性(82%);(3)血清IgM和IgG升高;(4)血清ALT、AST和ALP水平均升高;(5)小鼠脾脏T淋巴细胞群中CD8+细胞的比例和绝对数量显著增加,CD4+细胞未见明显变化,出现CD4+/CD8+比例倒置;(6)肝组织学观察到明显的胆管炎、胆汁淤积表现,损伤的胆管周围有大量CD4+和CD8+T淋巴细胞浸润,以CD8+T淋巴细胞为主,出现CD4+/CD8+比例倒置现象,偶见嗜酸性粒细胞;(7)血清IFNγ和IL-12p70水平升高;(8)实时PCR表明,AE2a,b-/-小鼠胆管细胞表现出在参与细胞氧化还原稳态和主要组织相容性复合体I类抗原呈递过程中的基因表达改变[17](表1)。胆管细胞中AE2的缺乏似乎扰乱了氧化还原平衡和蛋白质稳态,使胆管细胞易于被CD8+T淋巴细胞靶向攻击。
多项研究[18-20]观察到PBC患者的肝组织和外周血单核细胞中AE2 mRNA的表达减少,免疫组织化学显示PBC患者的胆管和肝细胞中AE2蛋白的表达降低;而且,从PBC患者分离培养的胆管细胞显示AE2存在明显缺陷。Banales等[21]研究发现来自PBC患者的胆管细胞中miR-506上调,与AE2 mRNA的3′UTR区域结合,防止蛋白质翻译,导致AE2活性降低,胆汁分泌功能受损。熊去氧胆酸是一种诱导碳酸氢盐富集的胆汁酸,可改善PBC患者的临床过程[22]。Prieto 等[23]研究PBC患者的肝活组织检查标本发现AE2 mRNA水平明显低于健康对照组,而使用熊去氧胆酸治疗后,AE2 mRNA水平升高至正常范围。AE2a,b-/-小鼠几乎所有淋巴细胞均具有Na+-HCO3-共转运体的酸化潜力,而CD8+T淋巴细胞却缺乏这种酸负荷机制,表现为碱化细胞内pH[24]。Mardones等[25]使用AE2a,b-/-小鼠的成纤维细胞观察在静息细胞内pH中碱性位移对cAMP信号和基因表达激活的影响,结果显示通过AE2的碳酸氢盐转运维持了培养的小鼠成纤维细胞内pH平衡,并揭示了cAMP在慢性碱化细胞反应中的作用。可见,AE2缺陷可能是PBC发病的重要机制之一。
1.4 ARE-Del-/-小鼠模型 ARE-Del-/-小鼠模型是通过敲出IFNγ基因3′非翻译区中具有162nt的腺嘌呤尿嘧啶富含元件(adenylate uridylate-rich element,ARE)而形成[26]。
该模型的生物表型特征包括:(1)20周龄时,雌性ARE-Del-/-小鼠比雄性小鼠的ALT、AST和总胆汁酸水平显著升高;(2)20周龄雌性ARE-Del-/-小鼠比雄性小鼠的肝脏门静脉区具有更加严重的淋巴细胞浸润、胆管损伤和肉芽肿形成;天狼猩红染色显示雌性ARE-Del-/-小鼠肝内轻度纤维化,而雄性小鼠纤维化不明显;(3)8~10周龄时,雌性ARE-Del-/-小鼠可检测到针对PDC-E2,BCOADCE2和OGDC-E2的自身抗体,主要是针对PDC-E2的抗体;(4)20周龄雌性ARE-Del-/-小鼠皮肤色素沉着明显增加,可能与胆管破坏导致胆汁流不足有关;(5)雌性ARE-Del-/-小鼠对趋化因子(IFNc诱导的单核因子、IFNc诱导型蛋白10和巨噬细胞炎性蛋白1β)和细胞因子(TNFα,IL-10和IL-13)具有更强的诱导作用;(6)从ARE-Del-/-转移到B6/Rag1-/-小鼠的CD4+T淋巴细胞诱导了中度的门静脉炎症和实质炎症;(7)肝脏基因表达的RNA测序显示上调基因与PBC胆管上皮细胞的基因表达特异性重叠,雌性小鼠中差异表达的基因具有更强的1型和2型IFN信号传导和淋巴细胞介导的免疫应答,表明IFNγ可能是推动PBC女性高发的重要因素[27](表1)。
该模型与人类PBC的不同点在于ALP无法测得、门静脉炎症和肝实质炎症主要由CD4+T淋巴细胞诱导,以及肝纤维化程度偏轻等。
2 化学或生物染毒类模型
2.1 2-辛炔酸偶联牛血清白蛋白(2-octynoic acid coupled to bovine serum albumin,2OA-BSA)小鼠模型
20A-BSA诱导的模型是指在完全弗氏佐剂的存在下,用2OA-BSA(100 μg/25 μl)腹膜内接种免疫小鼠,随后每个2周进行腹膜内接种加强1次以诱导模型。
2OA-BSA小鼠模型的血清和病理学特征:(1)特异性AMA阳性率100%且抗体水平明显升高;(2)用2OA-BSA免疫4周时小鼠血清中TNFα和IFNγ显著增加;(3)门静脉周围有大量淋巴细胞浸润,以CD4+和CD8+T淋巴细胞为主;(4)损伤胆管周围有轻度淋巴细胞或单核细胞浸润;(5)汇管区和肝实质中见上皮样肉芽肿形成;(6)肝实质中见少量肝细胞灶性坏死;(7)2OA-BSA免疫12周后,肝脏中CD4+T淋巴细胞比例降低,CD8+T淋巴细胞比例增高,CD4/CD8比值倒置;脾脏中CD4+T淋巴细胞和CD8+T淋巴细胞比例均降低,且CD4/CD8比值下降但无统计学差异[28](表1)。
不同于人类PBC的血清和病理学特征:(1)用2OA-BSA免疫24周时,未能检测到血清ALP等反映胆汁淤积的相关酶;(2)肝纤维化不明显;(3)未观察到肝内脂肪变性、嗜酸性粒细胞增多或胆汁淤积的改变;(4)其他组织如甲状腺、唾液腺、肺、肾、小肠和结肠等是正常的,未见免疫损伤,而PBC与多种肝外器官中的自身免疫病变相关[28]。
2.2 聚肌苷酸聚胞苷酸(polyinosinic polycytidylic acid,poly Ⅰ:C)小鼠模型 polyⅠ:C是一种1型IFN诱导剂,可诱导产生自身免疫性胆管炎。polyⅠ:C以5 mg/kg腹腔内注射雌性C57BL/6小鼠,2次/周,持续24~28周,即可制成polyⅠ:C小鼠模型。
该动物模型的血清和病理学特征:(1)特异性AMA检出率80%~100%;(2)血清ALT和ALP水平升高;(3)polyⅠ:C注射后3~6 h血清IFNα水平达到高峰,后逐渐下降,24 h后几乎检测不到;(4)诱导IL-12p70、IL-10、IL-6、单核细胞趋化蛋白-1、IFNγ等促炎细胞因子;(5)汇管区有大量炎症细胞浸润,T淋巴细胞和B淋巴细胞是构成浸润细胞的主体;在T淋巴细胞中,CD4+和CD8+T淋巴细胞在肝内均有定位,而CD8+T淋巴细胞的定位更靠近胆管。(6)在注射4周后,小鼠出现唾液腺炎、胰腺炎和间质性肾炎[29](表1)。可见,IFNα单独或与其他细胞因子结合可能是诱导PBC发病的关键因素,该模型可用于分析PBC的早期细胞事件。
Ambrosini等[30]观察2OA-BSA与polyⅠ:C偶联免疫小鼠是否会改变疾病过程,结果加入polyⅠ:C后导致自身免疫性胆管炎严重恶化,浸润性CD8+T淋巴细胞显著增加,促炎细胞因子水平也显著升高。提示先天免疫在自身免疫性胆管炎自然史中发挥重要作用。
2.3 大肠杆菌免疫小鼠模型 通过静脉内注射大肠杆菌感染NOD.B6-Idd10/Idd18小鼠,可制作血清AMA阳性和严重胆管炎的动物模型,这可能与大肠杆菌肽序列中具有6~8个氨基酸残基与人类PBC的PDC-E2自身表位相同有关[31]。该模型的生物学特征表现为:(1)血清AMA滴度在大肠杆菌感染NOD.B6-Idd10/Idd18小鼠4周时达到峰值,然后逐渐下降;(2)肝组织学显示在感染大肠杆菌26周时,出现肝内门静脉周围炎症反应和肉芽肿形成;(3)胆管损伤轻重不一,轻者胆管几乎完整,有轻度的淋巴细胞聚集,重者胆管上皮细胞几乎完全消失;(4)胆管炎的形成不需要激活NK T淋巴细胞(表1)[11,32]。
虽然该模型针对PDC-E2的反应活性较其他PBC动物模型弱,但在大肠杆菌感染早期抗PDC-E2的启动足以破坏免疫耐受性,并产生PBC样的病理损伤[32]。这一点强调了微生物感染在自身免疫性胆管炎中作为主要或共存次要煽动事件的重要性。
3 小结
PBC是一种器官特异性自身免疫性疾病,其免疫耐受性受到损害,但机制不明[33]。PBC的易感性可能是遗传和环境因素的结合,而环境触发是导致PBC发生发展的重要因素[34]。尽管PBC与自身免疫现象密切相关,但对经典的免疫抑制剂治疗反应较差。自身免疫鼠模型有助于研究PBC疾病发病机制中的早期事件。
AMA的特异性反映了PBC疾病发生的诱导阶段[35]。AMA的自身抗原靶标主要包括PDC-E2、BCOADC-E2和OGDC-E2。对PDC-E2耐受性的丧失是发生PBC的起始关键事件。在PBC中,T淋巴细胞和B淋巴细胞的表位集中在PDC-E2的硫辛酰结合域,PDC-E2硫辛酸通过对硫辛酰二硫键的亲电攻击进行化学修饰,触发了对PDC-E2耐受性的破坏[36]。因此,理想的PBC动物模型应具备以下条件:(1)雌性高发;(2)发病年龄:中年;(3)发病受遗传因素和环境因素的影响;(4)特异性自身抗体:硫辛酰结构域作为显性抗原表位,血清AMA阳性;(5)免疫失调:肝脏和血液中出现PDC-限制性CD4+和CD8+T淋巴细胞;调节性T淋巴细胞减少;肝脏NK细胞数目增多;(6)病理改变:选择性胆小管破坏;胆管周围T淋巴细胞浸润;肉芽肿形成;最终导致肝纤维化/肝硬化[37]。
基因修饰类小鼠模型如dnTGFβ RⅡ小鼠、NOD.c3c4小鼠、AE2a,b-/-小鼠和ARE-Del-/-小鼠,这些小鼠模型具有一个共同点就是自发形成PBC的选择性特征,如产生特异性自身免疫抗体AMA。化学或生物染毒诱导的PBC模型如2OA-BSA小鼠和polyⅠ:C小鼠模型,这些小鼠免疫耐受性的丧失主要是通过化学或生物染毒来诱导的。上述小鼠模型虽在某些方面模拟了PBC的发病特征,但仍存在多方面不足,因缺乏人类PBC诊断的基本标准,这些小鼠模型目前尚未被完全接受。
事实上,PBC是多种因素共同作用导致的,单一的动物模型很难完全模拟其免疫病理生理机制[11],目前这些动物模型虽有不同的缺陷,但已提高了对PBC病因和主要免疫学途径的认识。今后随着更加理想的动物模型问世,必将促进对PBC发病机制的深入理解及其药物研制。
[1] WEBB GJ, HIRSCHFIELD GM. Primary biliary cholangitis in 2016: high-definition PBC: biology, models and therapeutic advances[J]. Nat Rev Gastroenterol Hepatol, 2017, 14(2): 76-78.
[2] CHEN CW, CHENG J, DOU XG, et al. Consensus on the diagnosis and management of primary biliary cirrhosis (cholangitis)(2015)[J]. J Clin Hepatol, 2015, 31(12): 1980-1988. (in Chinese)
陈成伟, 成军, 窦晓光, 等. 原发性胆汁性肝硬化(又名原发性胆汁性胆管炎)诊断和治疗共识(2015)[J]. 临床肝胆病杂志, 2015, 31(12): 1980-1988.
[3] GORELIK L,FLAVELL RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease[J]. Immunity, 2000, 12 (2): 171-181.
[4] OERTELT S, LIAN ZX, CHENG CM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptorⅡdominant-negative mice[J]. J Immunol, 2006, 177(3): 1655-1660.
[5] LI MO, WAN YY, SANJABI S, et al. Transforming growth factor-beta regulation of immune responses[J]. Annu Rev Immunol, 2006, 24: 99-146.
[6] ANDO Y, YANG GX, KENNY TP, et al. Overexpression of microRNA-21 is associated with elevated pro-inflammatory cytokines in dominant-negative TGF-beta receptor type Ⅱ mouse[J]. J Autoimmun, 2013, 41: 111-119.
[7] CHUANG YH, LIAN ZX, YANG GX, et al. Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor Ⅱ dominant-negative mouse model of primary biliary cirrhosis[J]. Hepatology, 2008, 47(2): 571-580.
[8] KAWATA K, YANG GX, ANDO Y, et al. Clonality, activated antigen-specific CD8(+) T cells, and development of autoimmune cholangitis in dnTGFbetaRⅡ mice[J]. Hepatology, 2013, 58(3): 1094-1104.
[9] KOARADA S, WU Y, FERTIG N, et al. Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain[J]. J Immunol, 2004, 173(4): 2315-2323.
[10] IRIE J, WU Y, WICKER LS, et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis[J]. The J Exp Med, 2006, 203(5): 1209-1219.
[11] WANG J, YANG GX, TSUNEYAMA K, et al. Animal models of primary biliary cirrhosis[J]. Semin Liver Dis, 2014, 34(3): 285-296.
[12] TERASAKI S, NAKANUMA Y, YAMAZAKI M, et al. Eosinophilic infiltration of the liver in primary biliary cirrhosis: a morphological study[J]. Hepatology, 1993, 17(2): 206-212.
[13] ROMERO MF, CHEN AP, PARKER MD, et al. The SLC4 family of bicarbonate (HCO(3)(-)) transporters[J]. Mol Aspects Med, 2013, 34(2-3): 159-182.
[14] LIU Y, YANG J, CHEN LM. Structure and function of SLC4 family [Formula: see text] transporters[J]. Front Physiol, 2015, 6: 355.
[15] ALPER SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers[J]. J Exp Biol, 2009, 212(Pt 11): 1672-1683.
[16] ALPER SL. Molecular physiology of SLC4 anion exchangers[J]. Exp Physiol, 2006, 91(1): 153-161.
[17] SALAS JT, BANALES JM, SARVIDE S, et al. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling [J]. Gastroenterology, 2008, 134(5): 1482-1493.
[18] PRIETO J, GARCIA N, MARTI-CLIMENT JM, et al. Assessment of biliary bicarbonate secretion in humans by positron emission tomography[J]. Gastroenterology, 1999, 117(1): 167-172.
[19] MEDINA JF, MARTINEZ A, VAZQUEZ JJ, et al. Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis[J]. Hepatology, 1997, 25(1): 12-17.
[20] MEDINA JF. Role of the anion exchanger 2 in the pathogenesis and treatment of primary biliary cirrhosis[J]. Dig Dis, 2011, 29(1): 103-112.
[21] BANALES JM, SAEZ E, URIZ M, et al. Up-regulation of microRNA 506 leads to decreased Cl-/HCO3-anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis[J]. Hepatology, 2012, 56(2): 687-697.
[22] CORPECHOT C, CARRAT F, BAHR A, et al. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis[J]. Gastroenterology, 2005, 128(2): 297-303.
[23] PRIETO J, QIAN C, GARCIA N, et al. Abnormal expression of anion exchanger genes in primary biliary cirrhosis[J]. Gastroenterology, 1993, 105(2): 572-578.
[24] CONCEPCION AR, SALAS JT, SARVIDE S, et al. Anion exchanger 2 is critical for CD8(+) T cells to maintain pHi homeostasis and modulate immune responses[J]. Eur J Immunol, 2014, 44(5): 1341-1351.
[25] MARDONES P, MEDINA JF, ELFERINK RP. Activation of cyclic AMP signaling in Ae2-deficient mouse fibroblasts[J]. J Biol Chem, 2008, 283(18): 12146-12153.
[26] HODGE DL, BERTHET C, COPPOLA V, et al. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice[J]. J Autoimmun, 2014, 53: 33-45.
[27] BAE HR, LEUNG PS, TSUNEYAMA K, et al. Chronic expression of interferon-gamma leads to murine autoimmune cholangitis with a female predominance[J]. Hepatology, 2016, 64(4): 1189-1201.
[28] WAKABAYASHI K, LIAN ZX, LEUNG PS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease[J]. Hepatology, 2008, 48(2): 531-540.
[29] OKADA C, AKBAR SM, HORIIKE N, et al. Early development of primary biliary cirrhosis in female C57BL/6 mice because of poly I∶C administration[J]. Liver Int, 2005, 25(3): 595-603.
[30] AMBROSINI YM, YANG GX, ZHANG W, et al. The multi-hit hypothesis of primary biliary cirrhosis: polyinosinic-polycytidylic acid (poly I∶C) and murine autoimmune cholangitis[J]. Clin Exp Immunol, 2011, 166(1): 110-120.
[31] BOGDANOS DP, BAUM H, GRASSO A, et al. Microbial mimics are major targets of crossreactivity with human pyruvate dehydrogenase in primary biliary cirrhosis[J]. J Hepatol, 2004, 40(1): 31-39.
[32] WANG JJ, YANG GX, ZHANG WC, et al. Escherichia coli infection induces autoimmune cholangitis and anti-mitochondrial antibodies in non-obese diabetic (NOD).B6 (Idd10/Idd18) mice[J]. Clin Exp Immunol, 2014, 175(2): 192-201.
[33] JIANG T, HAN Z, CHEN S, et al. Resistance to activation-induced cell death and elevated FLIPL expression of CD4+T cells in a poly Ⅰ: C-induced primary biliary cirrhosis mouse model[J]. Clin Exp Med, 2009, 9(4): 269-276.
[34] BOGDANOS DP, KOUTSOUMPAS A, BAUM H, et al. Borrelia Burgdorferi: a new self-mimicking trigger in primary biliary cirrhosis[J]. Dig Liver Dis, 2006, 38(10): 781-782.
[35] HIRSCHFIELD GM, GERSHWIN ME. The immunobiology and pathophysiology of primary biliary cirrhosis[J]. Annu Rev Pathol, 2013, 8(8): 303-330.
[36] WANG J, BUDAMAGUNTA MS, VOSS JC, et al. Antimitochondrial antibody recognition and structural integrity of the inner lipoyl domain of the E2 subunit of pyruvate dehydrogenase complex[J]. J Immunol, 2013, 191(5): 2126-2133.
[37] LYU J, TAO YY, LIU CH. The characteristics and progress of primary biliary cirrhosis animal model[J]. Chin J Gastroenterol Hepatol, 2009,18(7): 584-586. (in Chinese)
吕靖, 陶艳艳, 刘成海, 等. 原发性胆汁性肝硬化的动物模型特点与研究进展[J]. 胃肠病学和肝病学杂志, 2009, 18(7): 584-586.
引证本文:HAO J, LIU CH. A study on animal models of primary biliary cholangitis[J]. J Clin Hepatol, 2017, 33(11): 2117-2122. (in Chinese)
郝娟, 刘成海. 原发性胆汁性胆管炎的动物模型[J]. 临床肝胆病杂志, 2017, 33(11): 2117-2122.
(本文编辑:邢翔宇)
Astudyonanimalmodelsofprimarybiliarycholangitis
HAOJuan,LIUChenghai.
(InstituteofHepatology,ShuguangHospitalAffiliatedtoShanghaiUniversityofTraditionalChineseMedicine,Shanghai201203,China)
An ideal animal model of primary biliary cholangitis (PBC) plays an important role in the research on physiopathological mechanism and drug research and development. In recent years, great achievements have been made in animal models which can reflect the features of PBC, such as positive serum anti-mitochondrial antibody and immunopathological injury of the bile duct. There are various methods for the preparation of animal models of PBC at present, including chemical or biological exposure and gene knockout. However, these models cannot completely simulate PBC in humans, since they have different serological, immunological, and histopathological features. This suggests the complexity of pathological mechanisms of PBC, including gene specificity and environmental changes and helps us to understand the pathogenesis of events in the early stage of PBC, such as the break of immune tolerance and specific attack of biliary epithelial cells.
cholangitis, biliary; models, animal
R575.22
A
1001-5256(2017)11-2117-06
10.3969/j.issn.1001-5256.2017.11.013
2017-09-01;
2017-09-04。
郝娟(1985-),女,主要从事中西医结合防治慢性肝病研究。
刘成海,电子信箱:chenghailiu@hotmail.com。
