原发性胆汁性胆管炎的临床特征与治疗分层管理
2017-11-22陈瑜,韩英
陈 瑜, 韩 英
(空军军医大学附属西京医院 消化内科, 西安 710032)
专家论坛
原发性胆汁性胆管炎的临床特征与治疗分层管理
陈 瑜, 韩 英
(空军军医大学附属西京医院 消化内科, 西安 710032)
原发性胆汁性胆管炎(PBC)是一种异质性相对较强的疾病,不同患者的临床经过及表型可能存在较大差异。因此,必须给予长期的治疗和随访才能对患者进行准确的危险分层。通过观察PBC患者临床症状、检测指标的变化,对不同患者进行个体化的管理,才能使低风险的PBC患者得到规范化治疗,使高风险患者可能从各种精心设计的临床研究中获益。
胆管炎, 胆汁性; 临床方案; 疾病管理
原发性胆汁性胆管炎(primary biliary cholangitis,PBC)是一种慢性自身免疫性的胆汁淤积性疾病,如不给予早期、规范治疗可进展为肝硬化,甚至终末期肝病及肝癌。PBC的诊断主要基于乏力及瘙痒的临床表现,血清ALP和GGT升高,抗线粒体抗体(AMA)阳性。PBC的治疗管理目标为及时治疗,缓解症状,预防患者进入肝病终末期。PBC是一种异质性相对较强的疾病,不同患者的临床经过及表型存在较大差异,因此必须给予患者长期的个体化治疗和随访才能正确对患者进行危险分层。美国食品药品监督管理局已正式批准熊去氧胆酸(UDCA)和鹅去氧胆酸(CDCA)为PBC的治疗药物。目前,贝特类药物以及布地奈德用于PBC的治疗亦有探索。现综述PBC的疾病临床特点以及如何对患者进行合理分层治疗及管理。
1 PBC的临床经过及表型
随着人们对PBC认识的提高,尤其是AMA检测的广泛应用,越来越多的PBC患者在疾病早期(即肝硬化发生之前)得以诊断,且无需组织学证据的辅助[1-2]。PBC以女性发病为主,仅有5%~10%患者为男性。相比女性,PBC男性患者诊断时年龄更高(60岁 vs 55岁,P<0.001),治疗不应答的男性患者的肝癌风险也更高[3-5]。UK-PBC研究共纳入PBC患者2353例,发现PBC在较年轻女性中的发病率增加,约有25%的PBC女性患者<50岁,且发病年龄越小越不易达到生化应答标准[4]。瘙痒和乏力是PBC患者最常见的临床表现,但约60%的PBC患者在诊断时无明显症状[6]。
约5%~10%的PBC患者临床表现、生化检测及组织学检查结果均符合PBC表现,但AMA检测阴性。对于AMA阴性的PBC患者,部分抗核抗体包括抗PML、Sp100以及抗gp-210抗体被认为具有诊断价值。近年通过蛋白组学研究,发现了2个PBC自身抗原——kelch样蛋白(kelch-like,KLHL)12和重组人己糖激酶(hexokinase,HK)1,KLHL12抗体和HK1抗体对PBC均有较高特异性(>95%),其诊断价值尚有待进一步临床验证[7]。AMA具有诊断价值但无预后判断价值[4,8-9]。抗gp-210抗体具有预后判断价值,研究[10-11]表明基线时抗gp-210抗体活性提示患者肝衰竭/肝移植的风险增加6倍,是独立于生化应答的预测因子,可有助于早期前瞻性地辨别高风险患者。抗中性粒细胞抗体常见于结缔组织病中,在PBC患者出现抗着丝点抗体阳性与门静脉高压相关[10]。约60%的PBC患者同时具有肝外自身免疫表现,但其对肝病预后的影响尚不清楚[12]。
约8%~10%的PBC患者兼具AIH的特征[13],包括ALT升高>5×正常值上限(ULN),IgG升高>2×ULN,抗平滑肌抗体阳性以及组织学表现为中重度界板性肝炎等。上述特征可出现在初次诊断为PBC时,也可以在PBC诊断并治疗数年后出现。因此,指南推荐经UDCA规范治疗6~12个月之后生化应答不佳的患者应考虑是否存在AIH因素的影响。该类型患者被定义为AIH-PBC重叠综合征,需接受UDCA联合免疫抑制剂治疗。所有疑似AIH-PBC重叠综合征的患者均推荐行肝活组织检查帮助诊断,并确定界板炎症的程度。此外,AIH-PBC重叠综合征患者的肝抗原/肝胰抗原以及抗双链DNA抗体常表现为阳性,因此建议行上述检查辅助诊断[14]。
2 PBC的生化应答标准
血清胆红素水平是PBC疾病预后的重要指标,并被纳入多个预后评分系统中,胆红素异常提示疾病进入晚期[15-16],因此血清ALP水平是更适用的预后指标。目前一项最大的荟萃分析[8]纳入了4845例PBC患者,研究结果提示ALP与肝移植或死亡之间存在对数线性关系,在疾病早期即可提供预后信息,并与患者的随访时间、发病年龄、性别、疾病分期以及治疗状态无关。多项研究[17-20]表明ALP数值下降的百分比或正常化与临床改善密切相关(单独使用或与其他生化变量联合使用)。表1所示为目前各种生化应答标准,无论使用何种标准,PBC患者在经过1~2年UDCA规范治疗(13~15 mg·kg-1·d-1)后如果达到应答标准,则临床预后明显改善。生化不应答患者的肝癌风险明显升高。2017年欧洲肝病学会指南也指出,尽管传统上使用UDCA治疗一般在12个月后进行评价,实际上治疗6个月后评估的结果同样有效,患者可能获益更大。
由于一部分应答良好的患者仍有疾病继续进展并有发生各种不良事件的风险, 因此现有的生化应答标准还有待进一步完善。有证据表明,UDCA治疗后的肝静脉/门静脉压力梯度与PBC患者的非肝移植生存期相关,治疗2~3年后压力梯度下降达20%被视为风险降低。食管静脉曲张是预后不良因素,本中心提倡对PBC患者定期行内镜检查,评估门静脉高压及食管静脉曲张的情况。迄今为止,尚无可靠的非侵入性手段用于评估门静脉高压。APRI指数,即AST/PLT比值可用于预测门静脉压力及肝纤维化状况[21-22]。一项纳入了386例患者的多中心研究结果表明,基线和治疗1年后的APRI评分是非肝移植生存期的独立预测因子。基线水平APRI>0.54提示死亡或肝移植,即使UDCA治疗1年后获得生化应答,但APRI>0.54的患者预后明显劣于APRI达标患者,而生化无应答且APRI>0.54的患者预后最差[21]。本中心建议患者3~6个月定期随访,评价治疗效果及疾病进展状况。

表1 PBC患者对UDCA治疗的应答评估

评分系统治疗时间(月)评分参数UK-PBC 12 治疗12个月的胆红素、ALP和AST(或ALT)。基线时的Alb和PLT水平GLOBE 12 治疗12个月的胆红素、ALP、Alb和PLT。基线时的年龄
3 PBC的组织学及无创检查
PBC的诊断主要依靠临床症状以及血清学检测,但当诊断存疑时,肝活组织检查的作用是无可替代的,且肝活组织检查可以提供疾病活跃程度及严重程度等关键信息。当前有数个PBC的组织学评价系统[23-24],包括准确评估可能出现的界板炎症、胆管缺失、慢性胆汁淤积和纤维化程度等参数对于PBC患者的生化应答以及临床转归的预测均有重要作用(表2)。组织学检查是评估肝纤维化进展的金标准,但该检查存在有创性、局部组织不一定能够代表病变全貌等局限性,常规使用组织学检查对PBC患者进行分层比较困难。因此无创性评估手段越来越多地应用于评估肝纤维化的组织学分期[25-26]。目前至少有2项关于瞬时弹性成像评估PBC肝纤维化的大型队列研究,其中1项单中心回顾性研究[25]显示瞬时弹性成像是独立于生化应答的预后因子。肝硬度检测除了反映肝纤维化程度,也会受到肝外胆汁淤积的影响,因此不一定能够反映疾病全貌。有研究[27]表明,当PBC患者的肝硬度值(LSM)>9.6 kPa,或每年LSM增加>2.1 kPa时,提示临床发生肝移植、肝病相关死亡的风险升高。ELF评分增加1分,则肝相关不良事件发生的风险增加3倍。

表2 PBC活组织检查的病理特点及预后
4 PBC的药物治疗
口服UDCA的疗效已被大量临床研究广泛证实,国内外指南均推荐所有PBC患者终身服用UDCA(13~15 m·kg-1·d-1),可延缓组织学进展,明显延长患者非肝移植生存期。但仍有约40%患者对UDCA治疗应答不佳,其发生肝病不良事件的风险大大增加。
4.1 奥贝胆酸(OCA) OCA是核受体法尼酯X受体(FXR)的配体,通过激活FXR信号通路调节胆汁酸合成、分泌、转运及减毒等各个环节,从而减轻PBC患者的胆汁淤积状态。OCA的Ⅲ期临床研究[28]表明,在ALP>1.67×ULN和(或)TBil升高<2×ULN的患者中有明显疗效,欧洲肝病学会指南推荐对符合以上条件且对UDCA应答不佳的患者使用OCA治疗。OCA治疗的主要副作用是瘙痒,瘙痒也是导致临床研究中受试者退出的主要原因。指南推荐OCA治疗起始剂量为5 mg,根据患者的耐受性,6个月后加至10 mg。OCA有望改善PBC患者的长期预后,但目前该药每年花费为69 350美元,远高于患者可接受的成本效益(18 450美元/年),使大量患者短期内无法受益[29]。
4.2 布地奈德 部分PBC患者的肝活组织检查可发现肝界板炎症,针对该类患者应用布地奈德能够改善其生化学及组织学应答[30],UDCA联合布地奈德6~9 mg/d的疗效优于UDCA单药治疗。布地奈德是肝首过效应的糖皮质激素,口服后90%的药物可在肝内进行首过代谢,从而降低了激素所致的全身不良反应。随着肝硬化和门静脉高压的出现,布地奈德的药代动力学也随之发生改变,导致其在体内的水平增加,因此不适于PBC晚期患者的治疗。
4.3 贝特类药物 贝特类药物包括匹立尼酸(Wy-14 643)、氯贝特、苯扎贝特和非诺贝特等是过氧化物酶体增殖物激活受体家族PPARα的人工合成激活剂。近年研究表明PPARα通路可以从减少胆汁酸的合成,调节胆汁酸成分,降低其细胞毒性,促进胆汁的排泄,抑制炎症反应多种途径改善肝细胞胆汁淤积。有关苯扎贝特的报道多来自日本,有研究[31]显示,UDCA疗效不佳的PBC患者在加用苯扎贝特后血清ALP下降62.5%;另有报道称,苯扎贝特单药治疗或联合UDCA治疗均可改善PBC患者的胆汁淤积。最近一项Meta分析[32]纳入了6项来自日本的苯扎贝特随机临床试验,结果提示与无干预组或单用UDCA组相比,苯扎贝特治疗可降低PBC患者的ALP水平。与日本不同,美国及中国等国家使用苯扎贝特并不普遍,多使用非诺贝特进行血脂紊乱的治疗。近年来,也出现了许多尝试使用非诺贝特治疗PBC的报道。相关研究结果提示对于UDCA治疗不佳的PBC患者加用非诺贝特后可明显降低ALP、GGT、ALT、AST及胆固醇等生化指标,还能降低IgM水平。2015年一项荟萃分析[33]表明, 对于UDCA应答不佳的患者使用非诺贝特联合UDCA治疗后,可显著改善患者生化指标及IgM水平,完全应答率提高至44%~88%。整体来说PBC患者对非诺贝特治疗耐受良好。新型PPARα激动剂(MBX8025)治疗UDCA无应答PBC患者的安全性和有效性的临床试验(NCT2609048)已经结束,但尚未见结果报道。
4.4 PBC的膳食补充 骨质疏松是PBC常见的并发症,因此患者应定期检测血清维生素D水平,并补充钙片和维生素D。必要时也可以使用二磷酸盐,但由于其可引起胃炎及食管炎,因此不推荐存在食管静脉曲张的患者使用。由于胆汁淤积影响PBC患者的胆汁分泌,进而影响其脂肪吸收,并可能引起脂溶性维生素缺乏,因此PBC患者应考虑监测维生素A、D、E、K的水平,并进行相应补充[34-35]。
5 PBC的治疗管理流程
由于PBC的异质性,不同特点的PBC患者的转归预后大不相同。因此对患者进行及时合理的病情评估和风险分层有利于患者的个体化管理,高风险患者更加可能受益于各种临床研究。2016年《Hepatology》上提出的PBC患者入组临床研究流程可视为一项PBC患者的管理流程(图1)[36]。确诊PBC的患者经UDCA规范治疗12个月后,开始评估生化应答情况。但是如果患者符合:抗gp210阳性、基线LSM>9.6 kPa、发病年龄<50岁等危险因素,则考虑提前至治疗6个月后进行评估。生化应答不佳者,如转氨酶升高明显(>5×ULN),推荐行肝活组织检查以评价界板性肝炎的严重程度;无明显转氨酶升高者,可入组至相应临床研究。在生化应答完全的患者中,应进一步评价:APRI指数如>0.54,或每年LSM增长>2.1 kPa,或PBC评分系统如GLOBE评分增加,则仍应视为风险较高人群,可入组至相应的临床研究。
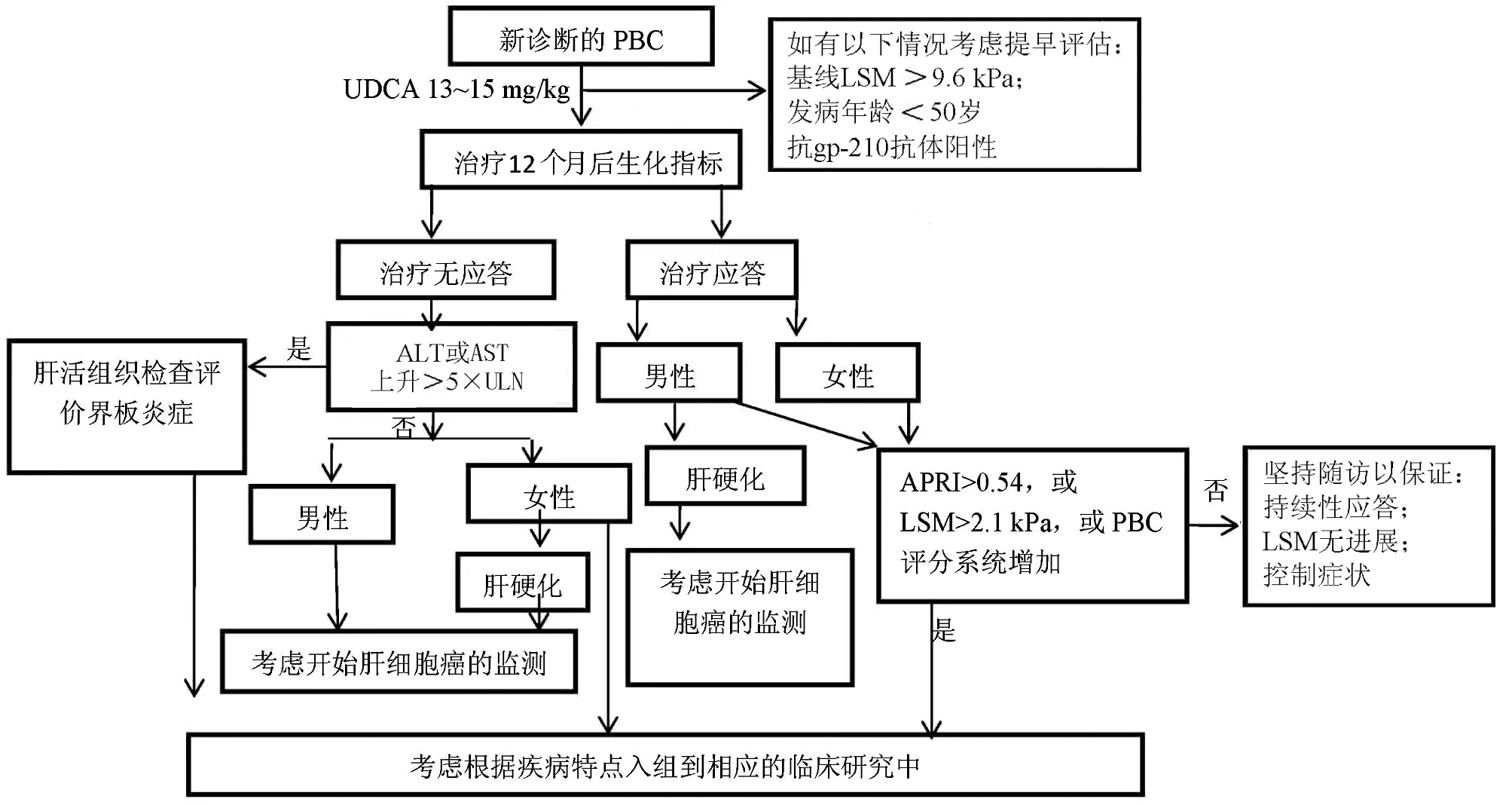
图1 PBC患者入组临床研究的洗程建议
肝移植后PBC的复发以及监测管理:UDCA治疗能够极大地改善PBC的病程及临床转归,但仍有约1/4的PBC患者最终由于终末期肝硬化、肝细胞癌及无法改善的瘙痒而转行肝移植手术。PBC患者移植术后的生存预后较好,据欧洲肝移植登记中心[37]报道,PBC患者移植后1年和5年生存率分别为90%和83%。患者移植后的生活质量取决于PBC的复发情况,相关研究结论可能受到样本量、随访时间及诊断复发的标准等因素影响,不同中心报道的复发率相差较大。在惯例进行肝活组织检查的中心所报道的复发率为10.9%~42.3%。近年一项纳入1241例PBC患者的荟萃分析[38]结果显示,随访时间中位数46.5个月时复发率为18%。目前被广泛接受的移植后复发PBC诊断标准是由Hubscher等[39]提出的:持续出现AMA、IgM升高以及组织学符合PBC的特征(淋巴浆细胞浸润、淋巴细胞聚集、上皮样肉芽肿及胆管损伤)。由于PBC病程进展缓慢,因此相对于丙型肝炎、AIH、原发性硬化性胆管炎复发的肝移植患者,PBC患者移植物丧失功能的风险最低。目前对于复发性PBC尚无标准治疗方案,观察性资料认为重新使用UDCA可明显改善肝酶学。有回顾性多中心研究[40]表明,预防性使用UDCA可明显降低PBC复发率。
6 小结
近年来人们对PBC的疾病特点及其疾病的异质性认识已有很大的提高。如何尽早准确识别出难治性PBC患者并予以针对性治疗是PBC诊治中的关键问题,相关问题尚需大量的研究探讨,尤其是前瞻性收集临床生化样本,并与配对的长期临床随访资料共同分析,可能有助于难治性PBC的诊断及内在机制研究。少数患者尽管到达生化应答标准,但仍会发生各种肝病相关不良事件;或者应答良好的患者在一段时间后实验室检查指标再度上升,这类患者也是临床工作中应予重视的对象,其潜在机制及临床特点还有待进一步研究。只有通过观察PBC患者临床症状、检测指标的变化,对不同患者实现治疗分层化的管理,才能使低风险的PBC患者得到规范化治疗,使高风险患者可能从各种精心设计的临床研究中获益。
[1] European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases[J]. J Hepatol, 2009, 51(2): 237-267.
[2] LINDOR KD, GERSHWIN ME, POUPON R, et al. Primary biliary cirrhosis[J]. Hepatology, 2009, 50(1): 291-308.
[3] TRIVEDI PJ, LAMMERS WJ, van BUUREN HR, et al. Stratification of hepatocellular carcinoma risk in primary biliary cirrhosis: a multicentre international study[J]. Gut, 2016, 65(2): 321-329.
[4] CARBONE M, MELLS GF, PELLS G, et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid[J]. Gastroenterology, 2013, 144(3): 560-569.e7.
[5] CHEUNG A, LAMMERS WJ, HIRSCHFIELD GM, et al. Age, bilirubin and albumin, regardless of sex, are the strongest independent predictors of biochemical response and transplantation-free survival in patients with primary biliary cirrhosis[J]. J Hepatol, 2015, 62: s798.
[6] PRINCE M, CHETWYND A, NEWMAN W, et al. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: follow-up for up to 28 years[J]. Gastroenterology, 2002, 123(4): 1044-1051.
[7] de LISO F, MATINATO C, RONCHI M, et al. The diagnostic accuracy of biomarkers for diagnosis of primary biliary cholangitis (PBC) in anti-mitochondrial antibody (AMA)-negative PBC patients: a review of literature[J]. Clin Chem Lab Med, 2017. [Epub ahead of print]
[8] LAMMERS WJ, van BUUREN HR, HIRSCHFIELD GM, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study[J]. Gastroenterology, 2014, 147: 1338-1349.
[9] TRIVEDI PJ, BRUNS T, CHEUNG A, et al. Optimising risk stratification in primary biliary cirrhosis: AST/plateletratio index predicts outcome independent of ursodeoxycholic acid response[J]. J Hepatol, 2014, 60(6): 1249-1258.
[10] NAKAMURA M, KONDO H, MORI T, et al. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis[J]. Hepatology, 2007, 45(1): 118-127.
[11] ZHANG LN, SHI TY, SHI XH, et al. Early biochemical response to ursodeoxycholic acid and long-term prognosis of primary biliary cirrhosis: results of a 14-year cohort study[J]. Hepatology, 2013, 58(1): 264-272.
[12] FLOREANI A, FRANCESCHET I, CAZZAGON N, et al. Extrahepatic autoimmune conditions associated with primary biliary cirrhosis[J]. Clin Rev Allergy Immunol, 2015, 48(2-3): 192-197.
[13] BOBERG KM, CHAPMAN RW, HIRSCHFIELD GM, et al. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue[J]. J Hepatol, 2011, 54(2): 374-385.
[14] European Association for the Study of the Liver. EASL Clinical Practice Guidelines: the diagnosis and managementof patients with primary biliary cholangitis[J]. J Hepatol, 2017, 67(1): 145-172.
[15] LAMMERS WJ, HIRSCHFIELD GM, CORPECHOT C, et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy[J]. Gastroenterology, 2015, 149(7): 1804-1812.
[16] CARBONE M, SHARP SJ, FLACK S, et al. The UKPBC risk scores: derivation and validation of a scoring system for long-term prediction of end-stage liver disease in primary biliary cirrhosis[J]. Hepatology, 2016, 63(3): 930-950
[17] CORPECHOT C, ABENAVOLI L, RABAHI N, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis[J]. Hepatology, 2008, 48(3): 871-877.
[18] PARÉS A, CABALLERA L, RODÉS J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid[J]. Gastroenterology, 2006, 130(3): 715-720.
[19] CORPECHOT C, CHAZOUILLRES O, POUPON R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long-term outcome[J]. J Hepatol, 2011, 55(6): 1361-1367.
[20] KUMAGI T, GUINDI M, FISCHER SE, et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis[J]. Am J Gastroenterol, 2010, 105(10): 2186-2194.
[21] TRIVEDI PJ, BRUNS T, CHEUNG A, et al. Optimising risk stratification in primary biliary cirrhosis: AST/platelet ratio index predicts outcome independent of ursodeoxycholic acid response[J]. J Hepatol, 2014, 60(6): 1249-1258.
[22] HUET PM, VINCENT C, DESLAURIER J, et al. Portal hypertension and primary biliary cirrhosis: effect of long-term ursodeoxycholic acid treatment[J]. Gastroenterology, 2008, 135(50): 1552-1560.
[23] KAKUDA Y, HARADA K, SAWADA-KITAMURA S, et al. Evaluation of a new histologic staging and grading system for primary biliary cirrhosis in comparison with classical systems[J]. Hum Pathol, 2013, 44(6): 1107-1117.
[24] WENDUM D, BOELLE PY, BEDOSSA P, et al. Primary biliary cirrhosis: proposal for a new simple histological scoring system[J]. Liver Int, 2015, 35(2): 652-659.
[25] CORPECHOT C, CARRAT F, POUJOL-ROBERT A, et al. Noninvasive elastography-based assessment ofliver fibrosis progression and prognosis in primary biliary cirrhosis[J]. Hepatology, 2012, 56(1): 198-208.
[26] FLOREANI A, CAZZAGON N, MARTINES D, et al. Performance and utility of transient elastography and noninvasive markers of liver fibrosis in primary biliary cirrhosis[J]. Dig Liver Dis, 2011, 43(11): 887-892
[27] MAYO M, PARKES J, ADAMS-HUET B, et al. Prediction of clinical outcomes in primary biliary cirrhosis by serum enhanced liver fibrosis (ELF) assay[J]. Hepatology, 2008, 48(5): 1549-1557.
[28] NEVENS F, ANDREONE P, MAZZELLA G, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis[J]. N Engl J Med, 2016, 375(7): 631-643.
[29] SAMUR S, KLEBANOFF M, BANKEN R, et al. Long-term clinical impact and cost-effectiveness of obeticholic acid for the treatment of primary biliary cholangitis[J]. Hepatology, 2017, 65(3): 920-928.
[30] DHANDA A, LEE R, COLLINS P. Is primary biliary cirrhosis a steroid-sensitive autoimme disease?[J]. Hepatol Res, 2012, 42(6): 619-620.
[31] IWASAKI S, TSUDA K, UETA H, et al. Benzafibrate may have beneficial effect in pre-cirrhotic primary biliary cirrhosis[J]. Hepatol Res, 1999, 16(1): 12-18.
[32] RUDIC JS, POROPAT G, KRSTIC MN, et al. Bezafibrate for primary biliary cirrhosis[J]. Cochrane Database Syst Rev, 2012, 1: cd450091.
[33] GRIGORIAN AY, MARDINI HE, CORPECHOT C, et al. Fenofibrate is effective adjunctive therapy in the treatment of primary biliary cirrhosis: a meta-analysis[J]. Clin Res Hepatol Gastroenterol, 2015, 39(3): 296-306.
[34] PHILLIPS JR, ANGULO P, PETTERSON T, et al. Fat-soluble vitamin levels in patients with primary biliary cirrhosis[J]. Am J Gastroenterol, 2001, 96(9): 2745-2750.
[35] MAILLETTE de BWL, BEUERS U. Bile salts and cholestasis[J]. Dig Liver Dis, 2010, 42(6): 409-418.
[36] TRIVEDI PJ, CORPECHOT C, PARES A, et al. Risk stratification in autoimmune cholestatic liver diseases: opportunities for clinicians and trialists[J]. Hepatology, 2016, 63(2): 644-659.
[37] ADAM R, KARAM V, DELVART V, et al. Evolution of indications and results of liver transplantation in Europe. A report from the European Liver Transplant Registry (ELTR)[J]. J Hepatol, 2012, 57(3): 675-688.
[38] GAUTAM M, CHERUVATTATH R, BALAN V. Recurrence of autoimmune liver disease after liver transplantation: a systematic review[J]. Liver Transpl, 2006, 12(12): 1813-1824.
[39] HUBSCHER SG, ELIAS E, BUCKELS JA, et al. Primary biliary cirrhosis. Histological evidence of disease recurrence after liver transplantation[J]. J Hepatol, 1993, 18(2): 173-184.
[40] BOSCH A, DUMORTIER J, MAUCORT-BOULCH D, et al. Preventive administration of UDCA after liver transplantation for primary biliary cirrhosis is associated with a lower risk of disease recurrence[J]. J Hepatol, 2015, 63(6): 1449-1458.
引证本文:CHEN Y, HAN Y. Clinical features of primary biliary cholangitis and stratified treatment management[J]. J Clin Hepatol, 2017, 33(11): 2095-2100. (in Chinese)
陈瑜, 韩英. 原发性胆汁性胆管炎的临床特征与治疗分层管理[J]. 临床肝胆病杂志, 2017, 33(11): 2095-2100.
(本文编辑:邢翔宇)
Clinicalfeaturesofprimarybiliarycholangitisandstratifiedtreatmentmanagement
CHENYu,HANYing.
(DepartmentofGastroenterology,XijingHospitalAffiliatedtotheFourthMilitaryMedicalUniversity,Xi′an710032,China)
Primary biliary cholangitis (PBC) is a disease with relatively strong heterogeneity, and different patients may have great differences in clinical process and phenotypes. Therefore, long-term treatment and follow-up are necessary for accurate risk stratification of these patients. Observation of clinical symptoms of PBC patients, measurement of the changes in related indices, and individualized management for each patient helps to provide standard treatment for low-risk PBC patients and bring benefits to high-risk patients through well-designed clinical studies.
cholangitis, biliary; clinical protocols; disease management
R575.22
A
1001-5256(2017)11-2095-06
10.3969/j.issn.1001-5256.2017.11.009
2017-08-06;
2017-08-31。
陈瑜(1979-),女,主治医师,副教授,主要从事自身免疫性肝病的诊断治疗及发病机制研究的工作。
韩英,电子信箱:hanying@fmmu.edu.cn。
