新型噻吩并吡啶双杂环化合物的合成研究
2017-11-14赵长阔王先恒杨福红保玉娇
赵长阔,王先恒,高 磊,杨福红,保玉娇,李 婵
(遵义医学院药学院,贵州 遵义563099)
新型噻吩并吡啶双杂环化合物的合成研究
赵长阔,王先恒*,高 磊,杨福红,保玉娇,李 婵
(遵义医学院药学院,贵州 遵义563099)
以市场易得的苯乙酮、噻吩-2-甲醛为原料,经过羟醛缩合,得到的α,β-不饱和酮化合物2与2-氰基乙硫乙酰胺发生迈克尔加成反应环合生成2H-吡啶硫酮3;最后在碱性条件下,与溴代苯乙酮一步合成新型噻吩并吡啶双杂环化合物1,总收率为25.4%。该方法具有原料易得,操作简便,产物收率高等优点,可推广合成至该类其它取代的吡啶双杂环衍生物。
苯乙酮;噻吩-2-甲醛;一锅法;吡啶双杂环衍生物
促黄体激素释放激素 (Luteinizing hormone releasing hormone,LHRH),是由下丘脑分泌的一种激素,结构为包含10个氨基酸的短肽Pyro-Glu-His-Trp--Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2,通过调控垂体内促黄体生成激素(LH)和促卵泡激素(FSH)的分泌,来调控人体激素水平及生殖功能[1-2]。LHRH的类似物临床上主要用于闭经、不孕症、子宫内膜异位症等妇科疾病以及激素依赖型肿瘤等治疗[3-5]。由于这些特殊癌症细胞增殖对生长激素有一定程度的依赖性,因此抑制性腺机能可直接或间接抑制癌细胞[6-7]。
促黄体激素释放激素类似物临床应用较多的还是肽类生物制品,化学合成的非肽类小分子药物在这一适应症领域还不多,例如用于治疗黄体功能不足的氯米芬 (Clomiphene).在PCT专利WO2000061586中公开了一类具有促黄体激素释放激素激动效应的噻吩并吡啶双杂环的化学结构,化合物1是此类结构的代表。选取人体绒膜促性腺激素HCG作为阳性对照,此类噻吩并吡啶双杂环结构能够较好地诱导雌性未成熟的小鼠排出成熟的卵母细胞[8]。
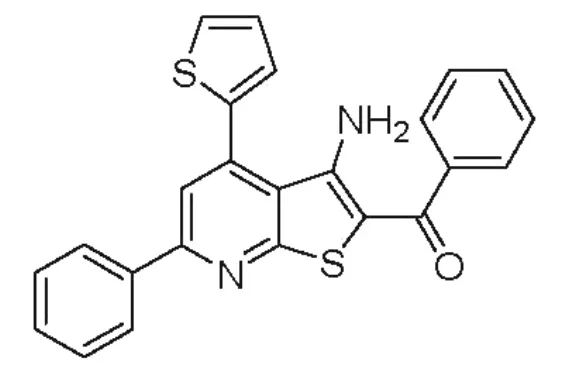
图1 化合物1
为深入研究此类化合物的生物活性,本项目在参照Gerritsma G.G.公开的合成通法[8],以苯乙酮和噻吩-2-甲醛为起始原料,通过羟醛缩合,迈克尔加成,亲和取代及成环反应四个步骤合成目标产物1时,发现亲核取代和成环反应可通过“一锅法”进行,最后经过优化反应条件后,以较高收率合成了化合物1,HPLC纯度在96.5%以上。
1 材料与方法
1.1 材料
苯乙酮、噻吩-2-甲醛、2-氰基乙硫乙酰胺、2-溴-1-苯基乙酮 (分析纯,国药集团);氢氧化钾、冰醋酸、石油醚、乙醚、氯仿、二氯甲烷、乙酸乙酯、四氢呋喃(分析纯,重庆川东化工);KOH、Na2CO3(分析纯,上海化学试剂厂);柱层析用硅胶(粒度200~300目,青岛海洋化工厂)、硅胶薄层板(青岛海洋化工厂);蒸馏水(自制)。
仪器采用81-2型暗箱三用紫外分析仪 (上海司乐仪器有限公司);Varian 400 MHz核磁共振波谱仪(TMS作内标,安捷伦科技有限公司);LC-10ATVP型高效液相色谱仪(日本岛津公司);液相色谱串联质谱联用仪(AB SCIEXL distribution)。
1.2 方法
1.2.1 (3-氨基-6-苯基-4-(噻吩-2-基)噻吩并[2,3-b]吡啶-2-基)(苯基)甲酮(1)的合成路线
Gerritsma G.G.[8]及Sharanin Y.A.等[9-10]报 道了以苯乙酮,噻吩-2-甲醛为原料,经过羟醛缩合(Aldol condensation),得到的 α,β-不饱和酮化合物2;再与2-氰基乙硫乙酰胺发生迈克尔加成反应(Michael addition)环合生成1,2-二氢吡啶3;与溴代苯乙酮发生亲核取代反应得到硫醚4,最后在强碱条件下,例如醇钠,成环生成目标产物1。反应路线参考图2。

图2 合成路线[8-10]
在我们尝试用这种图2所示合成路线[8-10]制备目标化合物时发现,化合物3和溴代苯乙酮在NaOEt等强碱条件下,可“一步”或“一锅”合成得到目标产物1,而无需分离中间产物4,收率高达85%。见图3和表3。

图3 “一步”法合成化合物1
1.2.2 (E)-1-苯基-3-(噻吩-2-基)-2-丙烯-1-酮(化合物2)的制备
将苯乙酮(12.0 g,100 mmol)和噻吩-2-甲醛(11.8 g,100 mmol)的混合物溶于甲醇(280 mL)中,加入15%氢氧化钾水溶液(48 mL)。搅拌反应2 h后,产物从反应液中沉淀出来。加入约20 mL的冰醋酸;减压蒸除溶剂后,加入蒸馏水(500 mL)和乙酸乙酯(500 mL)。分离有机相,水相用乙酸乙酯(200 mL)萃取;合并有机相用盐水(200 mL)洗涤。减压蒸除溶剂后得残余物,通过硅胶柱色谱法(溶剂:石油醚/乙酸乙酯,50:1至10:1梯度洗脱)纯化,得到(E)-1-苯基-3-(噻吩-2-基)-2-丙烯-1-酮的目标产物13.6 g,收率63%,为浅黄色固体。1H NMR(300 MHz,DMSO-d6)δ(ppm):8.15(d,J=4.8 Hz,2H),7.93(d,J=15 Hz,1H),7.79(d,J=4.8 Hz,1H),7.52~7.71(m,5H),7.20(t,J=3.9 Hz,1H).LC-MS:[M+1]=215.1;HPLC(214 nm/254 nm)=95.8%/91.6%。1.2.36-苯基-3-氰基-4-(噻吩-2-基)-2-硫代-1,2-二氢吡啶(化合物3)的制备
将(E)-1-苯基-3-(噻吩-2-基)-2-丙烯-1-酮(2.14 g,10 mmol)和2-氰基乙硫乙酰胺(1.0 g,10 mmol)溶于乙醇(30 mL),然后加入两滴哌啶。将混合物搅拌回流3 h。 滤出生成的固体产物,用冷乙醇(20 mL)洗涤;得到1.46 g(47.4%)6-苯基-3-氰基-4-(噻吩-2-基)-2-硫代-1,2-二氢吡啶,为橙黄色固体粉末。1HNMR(300 MHz,DMSO-d6)δ (ppm):7.87~7.96(m,4H),7.49~7.51(m,3H),7.27(t,J=4.2 Hz,1H),7.15(s,1H).LCMS:[M+1]=295.0;HPLC(214 nm/254 nm)=98.8%/98.0%。
1.2.4 (3-氨基-6-苯基-4-(噻吩-2-基)噻吩并[2,3-b]吡啶-2-基)(苯基)甲酮(化合物(1)制备
将6-苯基-3-氰基-4-(噻吩-2-基)-2-硫代-1,2-二氢吡啶 (1.46 g,4.9 mmol)、2-溴-1-苯基乙酮(0.99 g,4.9 mmol)与甲醇钠(将149 mg,6.0 mmol新鲜金属钠溶于40 mL甲醇溶液,现场制备得到)中在甲醇溶剂中回流加热3 h。过滤得到的固体产物,并用甲醇60 mL洗涤;得到(3-氨基-6-苯基-4-(噻吩-2-基)噻吩并[2,3-b]吡啶-2-基)(苯基)甲酮(化合物1)1.72 g;收率85.2%;为淡黄色粉末。1HNMR(300 MHz,DMSO-d6)δ(ppm):8.22(m,8.21~8.23,2H),7.97(d,J=4.8 Hz,1H),7.91(s,1H),7.79(d,J=7.2 Hz,2H),7.52~7.62(m,7H),7.35(t,J=4.8 Hz,1H),7.27(br,2H).LC-MS:[M+1]=413.0.HPLC(214 nm/254 nm)=96.5%/99.1%
(2)HPLC测定条件
层析柱:Agela Venusil MP C-18(5 μm,4.6×150 mm);流动相:梯度洗脱,70%CH3CN(0.05%TFA)、30%H2O(0.05%TFA)、100%CH3CN(0.05%TFA);流速:1.0 mL/min;检测波长:214 nm/254 nm。
2 实验条件的优化
我们重点对最后一步:一锅法合成吡啶衍生物1进行了工艺优化,从碱催化剂选择,反应温度,回流时间及溶剂选择等四方面进行优化探索。

2.1 不同的碱催化剂对收率的影响
碱催化剂选择对反应收率有较大影响:为促进反应进行,碱催化剂投料可相对底物略微过量,将摩尔投料比设为n(二氢砒啶3):n(溴代苯乙酮) :n(碱催化剂)=1:1:1.2,我们发现不同强度的碱催化剂对这步反应的收率影响较大。如果碱性太弱,如Na2CO3,几乎没有产物生成。

表1 不同碱催化剂对化合物1收率的影响Tab.1 Effect of different base on the yield of the compound 1
2.2 反应溶剂对收率的影响
将反应温度设定为回流温度,投料比设为n(二氢砒啶3) ∶n(溴代苯乙酮)∶n(EtONa)=1∶1∶1.2,其他操作如1.2.1所述,选取THF、CH3OH、EtOH、CH3CN以及1,4-二氧六环5种溶剂,考察反应溶剂对收率的影响,结果如表4所示。
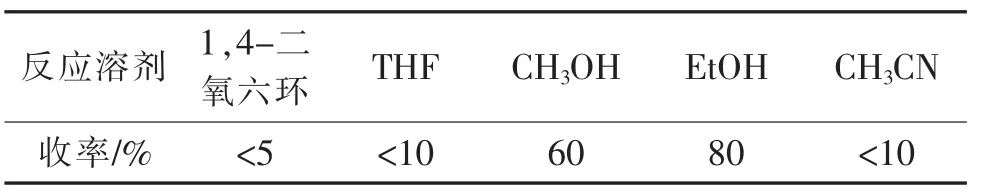
表2 不同反应溶剂对化合物1收率的影响Tab.2 Effect of different reaction solvent on the yield of the compound 1
结果表明:选取THF、CH3CN和1,4-二氧六环为反应溶剂,化合物1收率均较低;而在CH3OH和EtOH中化合物1收率较高。推测可能的原因是THF,CH3CN以及1,4-二氧六环为非极性非质子溶剂,对吡啶原料的溶解性较差,因此反应难以进行;而甲醇和乙醇是极性非质子溶剂,对两种底物的溶解性均很好,因此产物收率也高。
2.3 反应温度对收率的影响
选取以上优化条件,将反应时间设定为4 h,以乙醇为溶剂,选取室温、50℃和回流三阶段反应温度,考察反应温度对收率的影响趋势,结果如表3所示。

表3 不同温度对化合物1收率的影响Tab.3 Effect of different temperature on the yield of the compound 1
结果表明:温度越高,反应收率大幅提高。当在回流反应条件下,反应收率达到最大85%。当温度降低至0℃时,反应物不能获得足够活化能而几乎没有目标产物生成。
2.4 反应时间对收率的影响
选取以上优化条件,将反应温度设定为回流温度,以乙醇为溶剂,反应时间为2~8 h,考察反应时间对收率的影响,结果如表4所示。
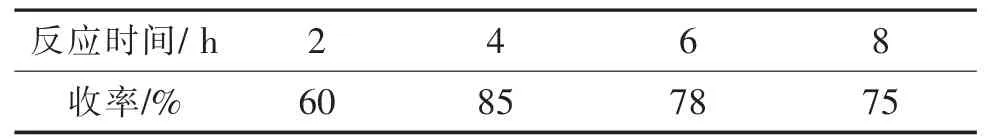
表4 不同反应时间对化合物1收率的影响Tab.4 Effect of different reaction time on the yield of the compound 1
结果表明,反应时间在2~4 h内,反应时间越长,反应收率越高;当反应时间达到4 h时,反应收率开始下降。原因是当主反应达到平衡后,开始向副产物方向进行。综合考虑,选择反应时间为4 h为宜。
因此,最佳反应条件为:110℃,EtOH为反应溶剂,n(二氢砒啶3)∶n(溴代苯乙酮)∶n(NaOEt)=1∶1∶1.2,反应4 h。通过放大实验(放大倍数10倍)三次,产品收率分别为85.4%、85.1%、85.1%,平均收率高达85.2%。
3 结论
本文以苯乙酮,噻吩-2-甲醛为原料,经过羟醛缩合,得到的α,β-不饱和酮化合物2;再与2-氰基乙硫乙酰胺发生迈克尔环合加成反应得到四氢吡啶3;最后在碱性条件下,与溴代苯乙酮一步合成新型的噻吩并吡啶双杂环化合物1,总收率为25.4%。该方法具有原料易得,操作简便,产物收率高等优点,可推广合成至该类其它取代的吡啶衍生物。
[1]高兴明,唐艳春,叶蕴华.促黄体生成激素释放激素类似物的研究进展[J].生物化学与生物物理进展,2000,27(5):500-504.
[2]Schally A V,Arimura A,Kastin A J.Gonadotropinreleasing hormone:one polypeptide regulates secretion of luteinizing and follicle-stimmmmmulating hormones[J].Science,1971,173(4001):1036-1038.
[3]马小玲,张炜.促性腺激素释放激素类似物在妇科疾病治疗中的应用[J].上海医药,2012,33(1):6-8.
[4]刘石萍,杨佳欣.促性腺激素释放激素类似物在妇科恶性肿瘤中的应用[J].国际妇产科学杂志,2012,39(5):514-517.
[5]戴军,沈周俊.促黄体生成素释放激素类似物、促黄体生成素释放激素拮抗剂及促黄体生成素释放激素新制剂对前列腺癌的疗效[J].上海医学,2009,32(12):1126-1128.
[6]Myburgh D B,Millar R P,Hapgood J P.Alanine-261 in intracellular loop III of the human gonadotropin-releasing hormone receptor is crucial for G-protein coupling and receptor internalization[J].Biochem J,1998,331:893-896.
[7]王卫芳.促黄体激素释放激素及其受体的研究进展[J].中国农学通报,2010,26(14):190-192.
[8]Gerritsma G G,Van Straten N C R,Adang A E P.Preparation of bicyclic heteroaromatic compounds as LH agonists:WO,2000061586[P].2000-10-19.
[9]El-Ossaily Y A.Convenient synthesis of some new indeno[1,2-b]pyridines and indeno[1,2-b]thieno[3,2-e]pyridine derivatives with potential biological activity[J].Phosphorus,Sulfur and Silicon and the Related Elements,2007,182(5):1109-1117.
[10]Sharanin Y A,Matrosova S V.Cyclization reaction of nitriles.LVI.Synthesis and conversion of substituted 6-aryl-4-(2-thienyl)-3-cyanopyridine-2(1H)-thiones[J].Zhurnal Organicheskoi Khimii,1996,32(8):1251-1255.
Research on the Synthesis of a New Thieno[2,3-b]pyridine Bicycle Compound
ZHAO Chang-kuo,WANG Xian-heng*,GAO Lei,YANG Fu-hong,BAO Yu-jiao,LI Chan
(School of Pharmacy,Zunyi Medical University,Zunyi,Guizhou 563099,China)
Starting from acetophenone and thiophene-2-formaldehyde,α,β-Unsaturated ketone compound 2 was synthesizes through Aldol condensation,the compound 2 reacted with 2-cyanoethylthioacetamide through Michael Cyclo-addition to afford 2H-pyridine-thione 3,which reacted with bromo-acetophenone under basic conditions to form the target product 1 containing the novel thieno[2,3-b]pyridine bicycle structure through a two-step reaction in One-pot,total yield reaches about 75%.The preparation method has advantages of easy availability of raw materials,simple operation and high yield of the product,and can be applied the other pyridine bicycle derivatives.
acetophenone;thiophene-2-carbaldehyde;one-pot synthesis;pyridine bicycle derivatives
1006-4184(2017)10-0016-04
2017-06-30
遵义医学院博士启动资金[2014]。
赵长阔(1971-),男,江苏淮安人,博士,副教授,研究方向:新药设计与合成、仿制药合成工艺改进等。E-mail:zhaochangkuo@163.com。
*通讯作者:王先恒,E-mail:wangxianheng01@163.com。
