硫改性镍催化剂丁烯-1双键临氢异构性能
2017-11-08王建强刘仲能周兴贵袁渭康
赵 多,王建强,刘仲能,周兴贵,袁渭康
1.华东理工大学 化学工程联合国家重点实验室,上海 200237;
2.中国石油化工股份有限公司上海石油化工研究院 绿色化工与工业催化国家重点实验室,上海 201208
硫改性镍催化剂丁烯-1双键临氢异构性能
赵 多1,2,王建强2,刘仲能2,周兴贵1,袁渭康1
1.华东理工大学 化学工程联合国家重点实验室,上海 200237;
2.中国石油化工股份有限公司上海石油化工研究院 绿色化工与工业催化国家重点实验室,上海 201208
将Ni/Al2O3催化剂浸渍于有机多硫化物(TPS-37)中制备得到硫改性Ni/Al2O3催化剂。采用X射线荧光光谱(XRF)、热重-质谱(TG-MS)联用、X 射线光电子能谱仪(XPS)和程序升温还原(TPR)等方法对催化剂进行表征,并考察了硫改性Ni/Al2O3催化剂上丁烯-1双键临氢异构性能。结果表明,硫改性Ni/Al2O3催化剂经高温氢气活化及甲苯萃取后,催化剂上硫含量基本不变。硫与催化剂之间存在较强的相互作用,使氧化镍在相对较低的温度下被H2还原;硫改变了Ni金属周围电子环境,从而显著改善催化剂的双键临氢异构性能。在反应温度60 ℃,氢压1.6 MPa,氢气和碳四烃物质的量之比(氢烃比)为0.013的条件下,硫改性Ni/Al2O3催化剂催化丁烯-1双键临氢异构反应,丁烯-1转化率可达92.6%,丁烯-2选择性为97.5%。催化剂稳定性实验结果表明,硫改性催化剂催化性能好,稳定性优良。
硫 镍 丁烯-1 临氢异构
裂解乙烯、催化裂化(FCC)及甲醇制烯烃(MTO)装置副产的碳四烃中均含有丁二烯、丁烯-1、丁烯-2和异丁烯等碳四烯烃,除少量用于异丁烯及丁烯-1分离外,其主要被用作燃气,而未被化工升值利用[1-3]。烯烃歧化(OCT/Meta-4)或裂解(OCC)增产丙烯及烷基化等技术的发展,以及对不含二烯烃的优质丁烯-2产品的旺盛需求,极大地推动了碳四烯烃的化工利用[4-7]。工业上已开始积极利用丁烯-1临氢异构制丁烯-2工艺。该工艺在脱除碳四烯烃中二烯烃的同时,可将丁烯-1转化成丁烯-2。
丁烯-1临氢工艺要求催化剂具有较好的双键异构选择性。Pd催化剂是最常用的选择加氢催化剂,在丁烯-1双键临氢异构反应中表现出较好的催化性能,丁烯-1异构化率大于 70%,丁二烯转化率大于98%,单烯烃收率大于100%[8]。Ni作为非贵金属催化剂,具有和贵金属Pd相近的双键异构活性[9],其成本明显低于贵金属,因而是很有发展潜力的双键异构催化剂。但单金属Ni催化剂上丁烯-1易发生加氢至丁烷,丁烯-2选择性较差。戴丹等[10,11]在负载型Ni金属催化剂上选择性地添加Mo或Ce等助活性组分,可提高Ni的分散度,还原后的 Ni也不易聚结,使催化剂活性得到改善。Hoffer等[12]对Ni进行部分预硫化处理可明显改善镍基催化剂的裂解汽油加氢选择性,抑制单烯烃及苯环加氢,提高汽油辛烷值。
本工作研究了S改性Ni/Al2O3催化剂的丁烯-1双键临氢异构性能,考察了S助剂对催化剂丁烯-1加氢活性和丁烯-2选择性的影响,并通过催化剂表征探讨了S助剂的作用机理。
1 实验部分
1.1 催化剂制备
按文献[13]方法制备NiAl2O3催化剂,记为Ni(O)。Ni(O)浸渍适量的二叔壬基多硫化物(TPS-37)得到预硫化 NiAl2O3催化剂 Ni(OS);Ni(O)经氢气高温还原并经微量氧缓慢钝化,得到一种外裹氧化镍壳层,内含镍晶粒的钝化型NiAl2O3催化剂Ni(RP);Ni(RP)浸渍适量的TPS-37,得到预还原硫化NiAl2O3催化剂Ni(RPS);氢气气氛下,160 ℃恒温活化处理Ni(RPS)催化剂8 h,即得到S改性NiAl2O3催化剂Ni(RPS-H)。
1.2 催化剂表征
Ni(RPS)和Ni(RPS-H)中S含量的测定:将催化剂置入索式提取器,用甲苯连续洗涤催化剂24 h,然后采用X射线荧光光谱(XRF)仪测定洗涤前后催化剂中的S含量;热重-质谱(TG-MS)分析采用德国耐驰公司生产的STA449型热重分析仪和QMS403C型质谱联用,分析条件为氢气气氛,流量50 mL/min,以10 ℃/min程序升温至180 ℃,质谱检测尾气中的H2S。
采用美国PerkinElmer公司生产的PHI5000C型X射线光电子能谱(XPS)仪测定Ni和S元素。测试条件为MgKa(hv为1 263.6 eV)X射线源,高压14.0 kV,功率300 W,通能93.6 eV。所有结合能数值均经过催化剂表面污染碳的C1s结合能(284.6 eV)进行样品荷电校正。
采用美国Micromeritics公司的AutochemII2920程序升温还原(TPR)装置进行氢气程序升温还原测定。催化剂预处理60 min后,在N2气流中降至室温,再通入氢气体积分数为10%的H2-Ar混合气,以10 ℃/min由室温升至800 ℃,热导池检测H2耗量。
1.3 催化剂性能评价
典型碳四原料组成(质量分数):异丁烷6.1%,正丁烷14.9%,反丁烯-2 18.1%,顺丁烯-2 12.3%,丁烯-1 47.5%,异丁烯1.0%和丁二烯0.047%。经过计量的碳四原料和氢气混合后进入固定床反应器,反应器内催化剂装填量30 mL,反应条件:反应温度60 ℃,氢气压力1.6 MPa,进料体积空速6 h-1,氢烃比(氢气和碳四烃物质的量之比)0.013。采用Agilent 7890D气相色谱对原料和产物组成进行在线分析。
丁烯-1双键临氢异构反应的主反应产物为丁二烯选择性加氢生成的丁烯-1及丁烯-1双键异构得到的丁烯-2,副产物为丁烯-1加氢生成的丁烷,主副反应如下:
主反应:

副反应:

丁二烯转化率(xBD)和丁烯-1转化率(x1B)分别由反应前后丁二烯和丁烯-1的质量分数变化计算得到,丁烯-2选择性(s2B)按下式计算。

2 结果与讨论
2.1 催化剂上硫含量分析
由于Ni(RPS)催化剂在使用前需于160 ℃氢气氛下活化8 h,而有机多硫化物在此温度下会发生分解,因此需考察经160 ℃高温处理后得到的Ni(RPS-H)催化剂上的S含量;H2S是镍临氢异构催化剂的毒物,可引起金属活性中心中毒失活,Ni(RPS-H)催化剂上的S在临氢反应过程中流失或生成H2S气体,将会影响催化剂的临氢异构性能。采用XRF法和TG-MS联用考察了催化剂上S含量的变化情况。表1为甲苯洗涤前后,Ni(RPS)和Ni(RPS-H)催化剂上S含量变化结果。由表可看出,经高温处理和甲苯萃取后,Ni(RPS)和Ni(RPS-H)催化剂上S含量未发生明显变化,在检测误差范围内。由此可推测,S与催化剂间存在较强相互作用,其在催化剂表面不是弱的物理吸附,而是发生了化学吸附。

表1 甲苯萃取前后催化剂上S含量的分析结果Table 1 S content of Ni(RPS) and Ni(RPS-H) catalysts before and after extraction of toluene
氢气气氛下,TG-MS联用分析结果见图1。由图可看出,温度在20~180 ℃时,催化剂的失重量约4%,可能是脱水和有机多硫化物分解导致的失重;质谱图上只在起始阶段出现H2S信号,在升温至160 ℃及以后的时间里都没有检测到H2S信号,表明Ni(RPS-H)催化剂上的S含量稳定。综上分析表明,Ni(RPS-H)催化剂不会因表面S含量的变化而引起催化性能的改变。

图1 氢气气氛下Ni(RPS-H)催化剂的TG-MS图谱Fig.1 TG-MS diagrams of Ni(RPS-H) treated in the hydrogen flow
2.2 硫改性镍基催化剂性能
在温度60 ℃,氢压1.6 MPa,氢烃比0.013条件下,考察了S和Ni质量比(S/Ni)对S改性Ni/Al2O3催化剂丁烯-1双键临氢异构性能的影响,结果如图2所示。由图可看出,Ni(RPS-H)具有较好的丁二烯加氢活性,丁二烯转化率达100%;采用未添加S的镍催化剂,丁烯-1转化率为12%,丁烯-2选择性为75%,部分烯烃加氢至烷烃;采用添加了S的催化剂后,丁烯-1的转化率和丁烯-2的选择性均得到了提高,且随着S含量增加,丁烯-1转化率及丁烯-2选择性升高明显,当S/Ni比为0.1时,丁烯-1转化率达到92.6%,丁烯-2选择性达97.5%;进一步增加S含量,丁烯-1转化率降低,丁烯-2选择性变化不大。上述结果表明,镍基催化剂上S及其含量是影响催化剂双键临氢异构性能的重要因素。不含S或S含量过低,部分烯烃加氢至烷烃,丁烯-2选择性差,但S含量过高,催化剂活性下降。
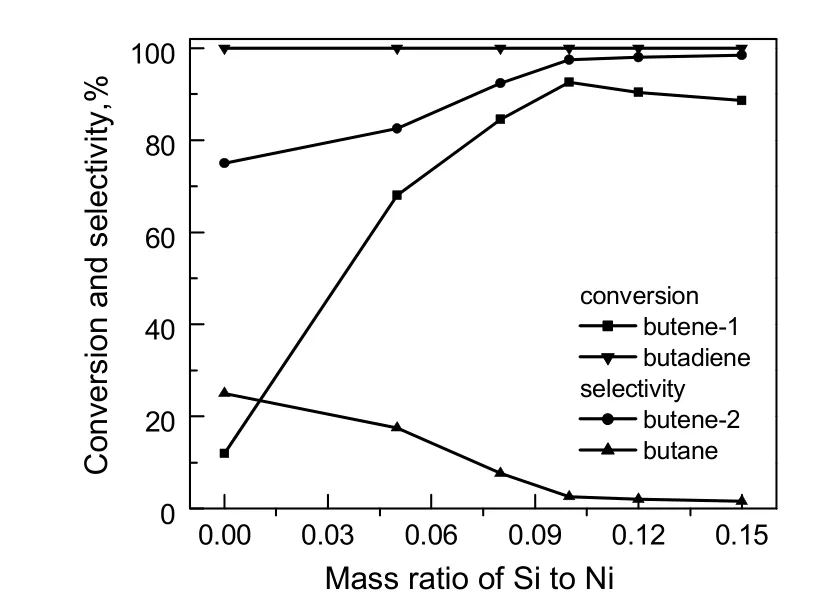
图2 S/Ni对催化剂催化性能的影响Fig.2 Effect of mass ratio of S to Ni on the catalytic performance of catalysts

图3 催化剂的H2-TPR图谱Fig.3 TPR profiles of catalysts
2.3 TPR分析
图3分别是Ni(O),Ni(OS),Ni(RP)和Ni(RPS)催化剂的H2-TPR谱图。Ni(O)催化剂上有两种NiO物种,低温耗氢峰(320 ℃)为游离态NiO的还原,而高温耗氢峰(470 ℃)为与载体作用较强的NiO的还原[14]。Ni(OS)催化剂上320和470 ℃的耗氢峰消失,而仅在256 ℃出现一个耗氢峰。说明引入多硫化物,催化剂上NiO的还原行为发生变化,在较低温度下即可还原成金属 Ni。Ni(RP)催化剂为经氢气还原和空气钝化后,金属Ni粒子的表面重新形成一种可低温(170 ℃)还原的氧化膜。Ni(RPS)催化剂的低温耗氢峰(154 ℃),与Ni(RP)催化剂的还原温度接近,应该是未吸附硫化物的表层NiO的还原,而高温耗氢峰(215 ℃),接近Ni(OS)的还原温度,应该是吸附硫化物的NiO的还原。实验结果表明,通过浸渍方法将多硫化物负载于催化剂表面后,多硫化物可与金属氧化物如Ni-O反应,发生化学吸附而形成Ni-O-Sz-CxHy化学键。在氢气存在下,多硫化物的分解与金属相态的转化同时进行,Ni-O-Sz-CxHy可在较低温度下还原为金属Ni。
2.4 S 2p3/2及Ni 2p3/2的XPS分析
S在金属表面的化学吸附既会阻碍反应分子接近活性位(几何效应),又会形成金属-硫键,改变金属位的电子特性(电子效应)[15]。
Barrio等[16,17]发现,有机硫化物(Sz)、有机硫化物的分解物(Sz-)及S2-3种形态S 2p3/2电子结合能分别为163.8,163.3和161.6 eV。图4给出了Ni(RPS)和Ni(RPS-H)催化剂上S 2p3/2 的XPS表征结果。由图可见,Ni(RPS)催化剂表面S 2p3/2的电子结合能163.6 eV,与研究[15,16,18]报道的Sz的结合能一致,表明S浸渍、吸附过程并没有改变该硫化物的电子结构。而Ni(RPS-H)催化剂表面S 2p3/2的电子结合能约161.9 eV,表明Ni(RPS-H)催化剂中S主要以S2-与Ni作用形成镍硫化物。Ni 2p3/2能谱中Ni0的电子结合能在852.6~853.3 eV,而Ni离子结合能因其配位离子和配位数的不同会有较大差别,但通常在854.0~857.5 eV[19,20]。据文献[21,22]报道,位于854.5 eV的谱峰是与载体相互作用较弱的自由NiO,位于855.4 eV(伴峰在861.7 eV)的谱峰归属于位于八面体配位的NiO,位于856.8 eV的峰是与载体相互作用较强或位于四面体配位的NiO和NiAl2O4组分。

图4 催化剂S 2p3/2的XPS图谱Fig.4 S 2p3/2 XPS spectra of the prepared catalysts

图5 催化剂Ni 2p3/2的XPS图谱Fig.5 Ni 2P3/2 XPS spectra of the prepared catalysts
图5给出了Ni(RP),Ni(RPS)和Ni(RPS-H)催化剂上Ni 2p3/2的XPS表征结果。由图可看出,Ni(RP),Ni(RPS)及Ni(RPS-H)催化剂表面均存在Ni0金属中心及缺电子的Ni2+,表明还原后的催化剂表面上除含有Ni0金属中心外,还有部分未被还原的金属氧化物。Ni(RP)催化剂表面Ni0的电子结合能为852.6 eV,Ni(RPS)催化剂表面Ni0的电子结合能依然为852.6 eV,而Ni(RPS-H)催化剂表面Ni0的电子结合能增至853.2 eV。表明S改性Ni/Al2O3催化剂上金属Ni电子云密度降低,有利于烯烃吸附的同时对H2的吸附解离能力下降,从而提高了丁烯-1双键临氢异构活性,有效地抑制了烯烃加氢至丁烷(见图2)。
2.5 催化剂稳定性实验
在温度60 ℃,氢压1.6 MPa,液相空速6 h-1,氢烃比0.013的工艺条件下,对S改性Ni/Al2O3催化剂进行了1 000 h稳定性实验,结果如图6所示。可看出,在运行期间内,催化剂表现出良好的活性、选择性和稳定性,丁二烯转化率100%,丁烯-1转化率大于92.5%,丁烯-2选择性保持在96.5%以上。

图6 硫改性Ni/Al2O3催化剂1 000 h稳定性实验结果Fig.6 Stability test results of S-modified Ni/Al2O3 catalyst for 1 000 h
3 结 论
a)采用有机多硫化物浸渍制备了S改性Ni/Al2O3催化剂,多硫化物化学吸附在催化剂表面,与氧化镍间存在较强相互作用,氢气升温还原时,硫化剂的分解与金属相态的转化同时进行,在催化剂表面形成镍硫化物。
b)S改性 Ni/Al2O3催化剂的部分活性位因形成镍硫化物电子云密度降低,从而有效地抑制催化剂的丁烯-1加氢活性,提高了双键异构产物丁烯-2的选择性。
c)S的添加及其加入量对催化剂性能影响显著。当S/Ni为0.1时,所制备的S改性Ni/Al2O3催化剂具有较优的催化性能。在温度60 ℃,压力1.6 MPa,氢烃比0.013的工况下,丁二烯转化率达100%,丁烯-1转化率大于92.6%,丁烯-2选择性大于96.5%,连续反应1 000 h,催化剂稳定性良好。
[1]张永军, 刘 剑, 汲永钢, 等.碳四馏分制低碳烯烃技术进展[J].化学工业, 2011, 29(10): 32-34.Zhang Yongjun, Liu Jian, Ji Yonggang, et al.Technical progress of olefins production from C4 fraction[J].Chemical Industry, 2011,29(10): 32-34.
[2]李 玲, 朱建宇, 吴久容, 等.混合碳四烃类的综合利用方案分析[J].广东化工, 2014, 41(11): 119-120.Li Ling, Zhu Jianyu, Wu Jiurong, et al.Analysis of integrated application over mixed C4 hydrocarbon[J].Guangdong Chemical Technology, 2014, 41(11): 119-120.
[3]王定博.炼厂碳四资源的利用途径[J].化工进展, 2014, 33(6): 1429-1434.Wang Dingbo.Utilization ways of refinery C4[J].Chemical Industry and Engineering Progress, 2014, 33(6): 1429-1434.
[4]戴 伟, 罗 晴, 王定博.烯烃转化生产丙烯的研究进展[J].石油化工, 2008, 37(5): 425-433.Dai Wei, Luo Qing, Wang Dingbo.Review of propylene production through olefins conversion[J].Petrochemical Technology, 2008,37(5): 425-433.
[5]张刘军, 徐春明, 高金森, 等.催化裂化C4烃催化转化增产丙烯[J].石油化工, 2005, 34(8): 714-718.Zhang Liujun, Xu Chunming, Gao Jinsen, et al.Catalytic conversion of catalytic cracking C4 hydrocarbon to propylene[J].Petrochemical Technology, 2005, 34(8): 714-718.
[6]徐海升, 李谦定, 马建平.碳四馏分选择加氢研究进展[J].现代化工, 2002, 22(8): 9-12.Xu Haisheng, Li Qianding, Ma Jianping.Advances in selective hydrogenation of C4 fraction[J].Modern Chemical Industry, 2002, 22(8): 9-12.
[7]张从良, 唐艳丽, 李学孟.异丁烷与丁烯烷基化反应催化剂的研究进展[J].郑州工业大学学报, 1999, 20(4): 25-28.Zhang Congliang, Tang Yanli, Li Xuemeng.Progress of research on catalysts for alkylation of isobutane and butene[J].Journal of Zhengzhou University of Technology, 1999, 20(4): 25-28.
[8]曹汉中, 牛春德, 王 昊, 等.丁烯-1临氢异构化催化剂及工艺技术开发[J].齐鲁石油化工, 2005, 33(1): 5-7.Cao Hanzhong, Niu Chunde, Wang Hao, et al.Catalyst and technology development butene-1 hydroisomerisation[J].Qilu Petrochemical Technology, 2005, 33(1): 5-7.
[9]Wells P B, Wilson G R.Butene isomerization catalyzed by supported metals in the absence of molecular hydrogen[J].Journal of Catalysis, 1967, 9(1): 70-75.
[10]戴 丹, 王海彦, 魏 民, 等.在Ni-Mo/Al2O3上催化裂化轻汽油的选择性加氢[J].辽宁石油化工大学学报, 2005, 25(2): 36-38.Dai dan, Wang Haiyan, Wei min, et al.Selective hydrogenation of FCC light gasoline over Ni-Mo/Al2O3[J].Journal of Liaoning University of Petroleum and Chemical Technology, 2005, 25(2): 36-38.
[11]李建卫, 黄星亮.低温选择性加氢镍催化剂的研究[J].石油化工, 2001, 30(9): 673-676.Li Jianwei, Huang Xingliang.Factors influencing the activity of Ni catalyst for low temperature selective hydrogenation[J].Petrochemical Technology, 2001, 30(9): 673-676.
[12]Hoffer B W, Devred F, Kooyman P J, et al.Characterization of ex situ presulfided Ni/Al2O3catalysts for pyrolysis gasoline hydrogenation[J].Journal of Catalysis, 2002, 209(1): 245-255.
[13]刘仲能, 吴晓玲, 王建强, 等.用于含少量丁二烯的丁烯-2临氢异构制丁烯-1的镍基催化剂: 中国, 101428225A[P].2007-11-07.
[14]Tsoncheva T, Linden M, Rosenholm J, et al.Nicke modified large pore mesoporous silicas as catalysts for methanol decomposition[J].Reaction Kinetics & Catalysis Letters, 2005, 86(2): 275-280.
[15]柴永明, 安高军, 柳云骐, 等.过渡金属硫化物催化剂催化加氢作用机理[J].化学进展, 2007, 19(z1): 234-242.Cai Yongming, An Gaojun, Liu Yunqi, et al.Transition metal sulfides hydrogenation catalysts: active phase structure and mechanism of the catalytic reaction[J].Progress in Chemistry, 2007, 19(z1): 234-242.
[16]Hoffer B W, Langeveld A D V, Janssens J P, et al.Stability of highly dispersed Ni/Al2O3catalysts: effects of pretreatment[J].Journal of Catalysis, 2000, 192(2): 432-440.
[17]Barrio V L, Arias P L, Cambra J F, et al.Evaluation of silica-alumina-supported nickel catalysts in dibenzothiophene hydrodesulphurisation[J].Applied Catalysis A: General, 2003, 248(1): 211-225.
[18]谢福中, 胡华荣, 乔明华, 等.噻吩在猝冷骨架Ni上吸附脱硫的XPS研究[J].高等学校化学学报, 2006, 27(9): 1729-1732.Xie Fuzhong, Hu Huarong, Qiao Minghua, et al.XPS study on adsorptive desulfurization of thiophene over rapidly quenched skeletal Ni[J].Chemical Journal of Chinese University, 2006, 27(9): 1729-1732.
[19]Poels E K, Beek W P V, Hoed W D, et al.Deactivation of fixed-bed nickel hydrogenation catalysts by sulfur[J].Fuel, 1995, 74(12):1800-1805.
[20]Guimon C, Horr N E, Romero E, et al.Characterization of the active sites of Ni-Si-Al sol-gel hydrogenation catalysts[J].Studies in Surface Science & Catalysis, 2000, 130: 3345-3350.
[21]Kirumakki S R, Shpeizer B G, Sagar G V, et al.Hydrogenation of naphthalene over NiO/SiO2-Al2O3catalysts: structure-activity correlation[J].Journal of Catalysis, 2006, 242(2): 319-331.
[22]Poncelet G, Centeno M A, Molina R.Characterization of reduced α-alumina-supported nickel catalysts by spectroscopic and chemisorption measurements[J].Applied Catalysis A General, 2005, 288(1/2): 232-242.
Performance of Sulfur Modified Catalysts for Hydroisomerization of 1-Butene
Zhao Duo1,2, Wang Jianqiang2, Liu Zhongneng2, Zhou Xinggui1,Yuan Weikang1
1.State Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China;
2.State Key Laboratory of Green Chemical and Industrial Catalysis, Shanghai Research Institute of Petrochemical Technology,SINOPEC, Shanghai 201208, China
The sulfur-modified Ni-based catalysts were prepared by the incipient wetness impregnation technique with the Ni/Al2O3in solution of organic polysulfide.The catalysts were characterized by X-ray fluorescence (XRF), thermogravimetry-mass spectrometry (TG-MS), X-ray photoelectron spectroscopy(XPS) and temperature-programmed reduction (TPR).The performance of sulfur-modified Ni/Al2O3catalyst on hydrogen isomerization of 1-butene double bond was investigated.The results showed that the sulfur content of the sulfur-modified Ni/Al2O3catalyst was almost unchanged after the activation of high temperature hydrogen and extraction with toluene.Since there was a strong interaction between the sulfur and the catalyst, nickel oxide was reduced by hydrogen at relatively low temperature.Sulfur changed the electronic environment around the Ni metal, thereby significantly improved the hydroisomerization of the catalyst at the double bond.The conversion up to 92.6%, selectivity of 2-butene 97.5% were obtained when sulfur modified Ni/Al2O3catalyst was used to catalyze the hydroisomerization of 1-butene under the operation conditions of temperature 60 ℃, hydrogen pressure 1.6 MPa and the molar ratio of H2to C4 0.013.The catalyst stability tests demonstrated that the sulfur- modified Ni/Al2O3performed excellent with good stability.
sulfur; nickel; 1-butene; hydroisomerization
TQ221.21+3;O643.32
A
1001—7631 ( 2017 ) 04—0305—07
10.11730/j.issn.1001-7631.2017.04.0305.07
2017-06-01;
2017-07-17。
赵 多(1978—),女,硕士。周兴贵(1966—),男,教授,通讯联系人。E-mail: xgzhou@ecust.edu.cn。
