高通量测序技术解析五粮液窖泥原核微生物群落结构
2017-11-03赵东郑佳彭志云吕学兰杨康卓张建敏
赵东,郑佳,彭志云,吕学兰,杨康卓,张建敏
(宜宾五粮液股份有限公司 技术研究中心,四川 宜宾,644000)
高通量测序技术解析五粮液窖泥原核微生物群落结构
赵东,郑佳*,彭志云,吕学兰,杨康卓,张建敏
(宜宾五粮液股份有限公司 技术研究中心,四川 宜宾,644000)
为清晰认识五粮液特殊窖泥微生态群落结构及功能,利用Illumina NextSeq 500高通量深度测序平台首次分析了五粮液不同窖泥原核微生物群落16S rDNA V4高变区序列;优化了QIIME程序并整理和统计样品序列数目和操作分类单元(OTUs)的数量,分析了不同窖泥中原核微生物群落的丰度、多样性;利用系统发育分析确定了优势菌群的系统发育地位。该研究结果表明,从五粮液窖泥中共获得7 555 164条高质量目标片段。在相同的测序深度条件下(300 000条),五粮液窖泥原核微生物主要由细菌和古菌构成,共检出22个门,52个纲,89个目,168个科,330个属原核微生物,细菌占92.3%,古菌占7.6%。厚壁菌门(Fimicutes)是所有样品的绝对优势菌群(85.6%),共检出74个属,Clostridium、Lactobacillus,Coprococcus,Caldicoprobacter,Soehngenia是丰度最高的细菌属。结合系统发育分析确定窖泥中优势菌群为Lactobacillus,Caldicoprobacter,Caloramater,Clostridium,Caloribacterium,Garciella,Eubacterium,Syntrophomonas,Sedimentbacter,Sporanaerobacter,Tissierella,Methanosarcina,Methanobacterium,Methanobrevibacter,Methanoculleus。五粮液窖泥中蕴藏了显著区别于其他浓香型白酒窖泥的原核微生物群落,尤其是群系复杂的厚壁菌门细菌,较高丰度的产己酸菌群和促进己酸生成的甲烷菌群。
五粮液;窖泥;原核微生物群落;高通量测序
五粮液,古称“重碧春、姚子雪曲、杂粮酒等”,是我国传统固态发酵白酒的典型代表。其独特的“五粮配方、包包曲制曲工艺、双轮底发酵、分层起糟、分层蒸酒、量质摘酒”等优秀的传统酿造技艺极大程度促进了我国白酒酿酒工业发展。五粮配方的续糟发酵是五粮液糟醅发酵的根本特征,长期不间断连续生产,尤其是糟醅的双轮底发酵,极大程度上提高了窖池内窖泥与糟醅的厌氧接触时间,糟醅与窖泥间营养物质、微生物的交换,形成了“以窖养糟、以糟促窖、糟窖互养”的良好互动局面。因此,确定窖泥微生物尤其是原核微生物的结构特征将有助于科学认识五粮液的酿酒微生物菌群代谢特征,为发酵过程代谢调控提供必要的依据。
窖池微生物群落的研究技术主要包括了传统分离培养技术、生物标记法和现代分子生物学免培养技术。20世纪90年代以来,吴衍庸等[1]基于传统分离培养技术剖析了五粮液老窖泥中厌氧微生物的群落结构,结果表明己酸菌和厌氧异养菌等随窖龄而增加,己酸菌、甲烷菌等随窖池上、中、下位置顺序递增。赵东等[2]分析了五粮液糟醅发酵过程中细菌(好氧、兼性厌氧)、酵母和霉菌的变化趋势。然而,传统培养法中由于培养基的高度选择性、微生物的独特生长特性等,迄今为止仅有微生物总数的0.1%~10%被检出[3],且此法操作过程费时费力,容易因培养基选择性、操作者的误差以及菌株生长条件需求差异等因素掩盖真实信息,难以定量表征群落多样性特征及胁迫条件的影响。进入21世纪,聚合酶链式反应(PCR)技术开始应用于浓香型白酒微生物群落结构的研究[4-6],一定程度上克服了传统分离培养技术的缺陷。但诸如PCR-DGGE、PCR-SSCP等手段的低分辨率(仅仅检出少数优势种群)、条带共迁移、条带DNA/RNA回收困难、无法定量确定条带比例等缺点,难以客观真实地定量反映窖泥微生物群落的演替规律。生物标记法,如磷脂脂肪酸(PLFA)[7-8]和麦角甾醇[9]等已应用于研究浓香型白酒微生物群落生物量的分布规律,但由于菌株间具有相似的PLFA组成等因素,菌株间PLFA相互干扰致使该法无法获得酿酒体系中微生物群落的组成信息。高通量测序技术是近年来快速发展并渗透至多学科的有效的分子生态学手段,无需分离纯培养单一菌株,提取体系基因组DNA后,直接在基因水平研究挖掘环境中微生物群落资源,同时,该方法体系兼具有通量高,覆盖率高等优点[10]。基于测序结果中reads数(序列条数)能进行不同操作分类单元(OTU)的数量的统计,经过OTU与微生物数据库的比对信息后即可获得相关微生物的分类信息,若在保证测序深度和测序质量的条件下,即可获得较为全面的进行微生物群落多样性评估信息。陶勇等[11]基于焦磷酸测序分析了四川某知名酒厂不同窖龄浓香型白酒窖泥中细菌和古菌的群落结构及多样性。邓杰等[12]利用焦磷酸测序技术分析了丰谷窖泥中原核微生物的结构组成。李克亚等[13]基于Illumina MiSeq测序分析了蜀之源和丰谷窖泥中原核微生物的群落结构。各个测序平台间由于测序原理的不同,获得样品测序深度完全不同(序列均在20 000条以内)。同时,样品间由于测序文库制备时的偏差致使测序深度具有差异性,直接在不同测序深度进行群落多样性评估势必存在一定弊端。因此,为了避免评估微生物群落丰度时出现偏差,需要在文库制备、测序平台选择、测序数据质量控制、生物信息学分析等方面进行层层技术把关。
为了获得全面的酿酒微生物群落结构特征以及其代谢特征,作为中国白酒领军企业的五粮液集团公司于2015年在国内酒类企业中率先购置了美国Illumina公司NextSeq 500高通量测序仪,该仪器具有灵活的测序类型(宏基因组、宏转录组、外显子、扩增子等)、高的测序通量(中通量20-40Gb,高通量30-120Gb数据量)以及较快的测序速度(12~30 h)等优势[14]。因此,本研究通过高通量深度测序策略进行五粮液窖泥原核微生物16S rDNA V4区序列进行测序;随后进行序列重采样,全面评估窖泥原核微生物群落组成及多样性。
1 材料和方法
1.1取样方法
所有样品均采自五粮液股份有限公司酿酒车间,窖池窖龄范围为15~50年。6口窖池分别位于不同的车间,所有窖池的生产工艺相同,均为续糟跑窖发酵工艺,且各窖池的原辅料品质类型在长期生产中均保持一致为选择窖池的标准。将窖池窖壁垂直中线处分为2个点,以黄水线为参考基准(距池底高度范围1.2~1.7 m),即中层和下层窖泥。采样方法见参考文献[5],每点采集3个平行样品,分别取100 g,混合均匀。6口窖池的样品编号分别从1#至12#(单数为各窖池的中层窖泥,双数为下层窖泥),其中1#-2#和3#-4#采集自车间一(窖龄约50年)、5#-6#和7#-8#采集自车间二(窖龄约20年)、9#-10#和11#-12#采集自车间三(窖龄约15年),样品置于真空袋中-20 ℃冷冻保存。
1.2仪器与试剂
NextSeq 500高通量测序仪,Illumina,美国;Qubit 2.0荧光定量仪,Thermo Fisher,美国;T100 梯度PCR仪,Bio-Rad,美国。
土壤DNA提取试剂盒(MP biomedicals,美国),KAPA HIFI Hotstart Readymix高保真热启动酶(KAPA Biosystems,美国),Qubit dsDNA HS Assay kit(Thermo Fisher,美国),引物由上海生工生物工程有限公司合成,Agencourt AMPure XP磁珠核酸纯化试剂盒(Beckman Coulter,美国),Nextera XT Index Kit v2 Set A 标签试剂盒(96 Indices,384 Samples,Illumina,新加坡),Nextera XT DNA Sample Preparation Kit 文库制备试剂盒(24 Samples,Illumina,新加坡),NextSeq 500/550 Mid Output Kit v2 中通量测序试剂盒(300 cycles,Illumina,新加坡)。
1.3基因组DNA提取及PCR扩增
采用土壤DNA提取试剂盒提取样品总DNA,具体操作步骤见试剂盒使用手册,然后用Qubit 2.0荧光定量仪测定其浓度,保证每个样品的DNA质量浓度大于10 μg/μL。
高通量测序区域采用细菌16S rDNA基因V4区域(Escherichiacoli位点515-806)的带有测序接头的特异性引物对Hyb515F(5’-T ̄C ̄G ̄T ̄C ̄G ̄G ̄C ̄A ̄G ̄C ̄G ̄T ̄C ̄A ̄G ̄A ̄T ̄G ̄T ̄G ̄T ̄A ̄T ̄A ̄A ̄G ̄A ̄G ̄A ̄C ̄A ̄G ̄G ̄T ̄G ̄C ̄C ̄A ̄G ̄C ̄M ̄G ̄C ̄C ̄G ̄C ̄G ̄G ̄T ̄A ̄A-3’)和Hyb806R(5’-G ̄T ̄C ̄T ̄C ̄G ̄T ̄G ̄G ̄G ̄C ̄T ̄C ̄G ̄G ̄A ̄G ̄A ̄T ̄G ̄T ̄G ̄T ̄A ̄T ̄A ̄A ̄G ̄A ̄G ̄A ̄C ̄A ̄G ̄G ̄G ̄A ̄C ̄T ̄A ̄C ̄H ̄V ̄G ̄G ̄G ̄T ̄W ̄T ̄C ̄T ̄A ̄A ̄T-3’),在每对引物的5’末端添加了能与Illumina测序flowcell表面结合以利于产生cluster的接头序列。PCR扩增具体反应程序如下:94 ℃预变性3min;94 ℃变性20 s,53 ℃退火25 s,72 ℃延伸45 s,共25个循环;72 ℃延伸5 min。利用磁珠法纯化PCR扩增产物(纯化方法见纯化试剂盒),再进行测序文库构建。
1.4测序文库构建
利用Nextera XT Index Kit v2 Set A 标签试剂盒和Nextera XT DNA Sample Preparation Kit文库制备试剂盒进行样品上机测序文库的构建,具体操作步骤见试剂盒操作手册。使用NextSeq 500高通量测序仪进行双端测序(Paired-end sequencing),产生读长为290 bp的序列。
1.5生物信息学分析
1.5.1 序列处理与分析
对测序原始数据通过bcl2fastq软件获得相应样品的原始序列,利用QIIME(Quantitative Insights into Microbial Ecology)对原始DNA序列进行过滤处理,除去嵌合体,并按照最大允许4个碱基错误为标准进行序列拼接,最终得到优化序列。按照97%相似性将优化序列划分操作分类单元OTUs(Operational Taxonomic Units)。对OTUs表进行重采样[15],使各样品获得一致的序列数,绘制稀释曲线(Rarefaction curve),计算Chao1丰度指数和Shannon多样性指数,得到微生物群落结构组成,其中重采样次数为2次。
Coverage指数的计算公式为:
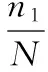
(1)
式中:n1代表只含有1条序列的OUT数目;N代表抽样中出现的总序列数目。
Shannon指数计算公式为:
H=-ΣPi×lnPi
(2)
式中:Pi为各种群物种数与样本总物种数之比。
Chao1指数计算公式为:
Chao1=OTUr+n1(n1-1)/2(n2+1)
(3)
式中:OTUr代表实际OUT数量;n1代表只有1条序列的OUT数量;n2代表只有2条序列的OUT数量。
利用QIIME软件计算样品Beta多样性距离矩阵,并根据Beta多样性距离矩阵进行主坐标分析(principal coordinate analysis,PCoA)。同时基于各样品中优势原核微生物的丰度,利用Canoco 4.5软件进行样品间关系的主成分分析(principal component analysis,PCA)。
1.5.2 优势序列系统发育分析
以QIIME中获得的丰度大于1%优势属OTUs序列为研究对象,通过RDP classifier(http://rdp.cme.msu.edu/classifier/classifier.jsp)与NCBI BLASTn中查出的同源性最高菌株序列为参考序列,利用MEGA 4.0软件以ClstralW进行序列比对,以最大相似距离估算系统进化矩阵,采用邻接法(Neighbor-Joining)构建优势OTU系统发育树,其中bootsrap次数为1 000[6]。
2 结果与分析
2.1测序结果
选择了五粮液6口窖池为研究对象,以黄水线为分界线,比较了干糟对应的中层窖泥和湿糟对应的下层窖泥中原核微生物群落结构。基于Illumina NextSeq深度测序平台测序,blc2fastq软件、QIIME程序拼接缝合序列后,12个窖泥获得了7 659 476条原始序列,质控后得到7 555 164条高质量目标片段(长度范围283~291 bp),其中序列数最少和最多的样品分别为7#窖泥(465 263条)和8#窖泥(801 675条)。
由于每个样品的测序深度不同,若要进行后续群落多样性比较,需要进行序列的重采样。对样品进行不同序列数采样(10 000条,20 000条,40 000条,100 000条和300 000条),使每个样品获得一致的序列数,计算样品的稀释曲线、覆盖度、多样性指数等参数。由表1可知,随着重采样序列数的增加,样品覆盖度增加,当达到30 000条时,覆盖度达99.5%,且各样品OTU稀释曲线区域平缓(图1),多样性指数包括Chao1、Shannon均随采样序列的增加而增加,表明300 000条的测序深度能够比较全面的反映本研究中窖泥原核微生物群落的多样性。

图1 丰度稀释曲线Fig.1 Rarefaction curves of pit muds

序列数覆盖度Chao1指数香农指数OTUs数量100000.966±01313±1155.51±0.04622±0.7200000.977±01812±1845.53±0882±5.7400000.984±02533±1845.55±01257±291000000.990±03972±1165.61±02032±373000000.995±05123±1715.62±03402±13
基于以上重采样序列,进行目标序列与数据库的比对,选择了3种常见的序列比对方法(uclust、blast和RDP),结果如表2所示,可知前2种比对方法在门、纲、科的数量差别不大,在属上有一定差异性,而RDP比对方法则具有明显优势。因此,将RDP作为序列比对的主要方法,共检出22个门,52个纲,89个目,168个科,330个属原核微生物。

表2 不同序列比对方法结果比较
2.2原核微生物群落多样性与群落组成
2.2.1 门水平的原核微生物群落多样性
本研究中,12个窖泥中原核微生物主要是由细菌和古菌构成,其中细菌占92.3%,古菌占7.6%。可见,细菌的丰度和多样性显著高于古菌,细菌为优势菌群。由表2可知,共检出19个门,包括:广古菌门(Euryarchaeota)、泉古菌门(Crenarchaeota)、酸杆菌门(Acidobacteria)、放线菌门(Acinobacteria)、拟杆菌门(Bacteroidetes)、衣原体门(Chlamydiae)、绿弯菌门(Chloroflexi)、蓝细菌门(Cyanobacteria)、芽单胞菌门(Gemmatimonadetes)、黏胶球形菌门(Lentisphaerae)、厚壁菌门(Fimicutes)、硝化螺旋菌门(Nitrospirae)、浮霉菌门(Planctomycetes)、变形菌门(Proteobacteria)、螺旋体门(Spirochaetes)、互养菌门(Synergistetes)、软壁菌门(Tenericutes)、疣微菌门(Verrucomicrobia)以及其他7种未命名门类(AC1、FBP、OP9、TM7、WS1、WWE)。优势门的丰度见图2,厚壁菌门(Fimicutes)在所有样品中均是绝对优势菌群(85.6%),其次为广古菌门(Euryarchaeota,7.6%)、拟杆菌门(Bacteroidetes,4.7%)、放线菌门(Acinobacteria,1.3%)和变形菌门(Proteobacteria)。1#~6#三对(1#和2#、3#和4#、5#和6#)窖泥中,中层窖泥中厚壁菌门细菌丰度低于下层窖泥,拟杆菌门和泉古菌门则反之。9#~12#两对(9#和10#、11#和12#)窖泥中厚壁菌门细菌的丰度则是中层高于下层,而泉古菌门的则是下层高于中层。

图2 门水平优势原核微生物菌群Fig.2 Dominantprokaryotic microbial communities at phylum levels in pit muds
2.2.2 属水平的原核微生物多样性分析
为进一步清晰的认识五粮液窖泥中重要的功能原核微生物菌群,本研究从属水平进行系统发育和统计分析。RDP分析显示绝大部分序列可归类到330属(细菌322属,古菌8属),其中丰度大于1%的属中细菌13个属(Lactobacillus-OTU975918,Caldicoprobacter-OTU4319602,Caloramater-OTU584933,Clostridium-OTU4342297,Thermoanaerobacterium-OTU4411374,Garciella-OTU742140,Coprococcus-OTU585881,Dorea-OTU349343,Rummincoccus-OTU771083,Syntrophomonas-OTU4390692,Sedimentbacter-OTU805522,Sporanaerobacter-OTU4312474,Tissierella-OTU4320991),古菌4属(Methanosarcina-OTU264516,Methanobacterium-OTU105600,Methanobrevibacter-OTU22694,Methanoculleus-OTU283331),各优势菌属的丰度如图3所示。可见,部分窖池的中层窖泥中原核微生物群落与下层微生物群落结构组成具有一定相似性(如1#和2#、11#和12#),而部分窖池的差异却很大(5#和6#、7#和8#、9#和10#)。部分窖池的中层窖泥中的甲烷菌群丰度显著高于下层(1#>2#,3#>4#,5#>6#、11#>12#),而7#和8#中几乎没有检出甲烷菌群,9#中甲烷菌的丰度则小于10#。1#与2#、11#与12#间差异最明显的是Methanobacterium,3#与4#间主要是Garciella、Caloramator、Ruminococcus和Sporanaerobacter的丰度差别,5#与6#、7#与8#、9#与10#两两间的差异十分显著,主要差异的微生物种属包括Lactobacillus、Methanobacterium、Ruminococcus。
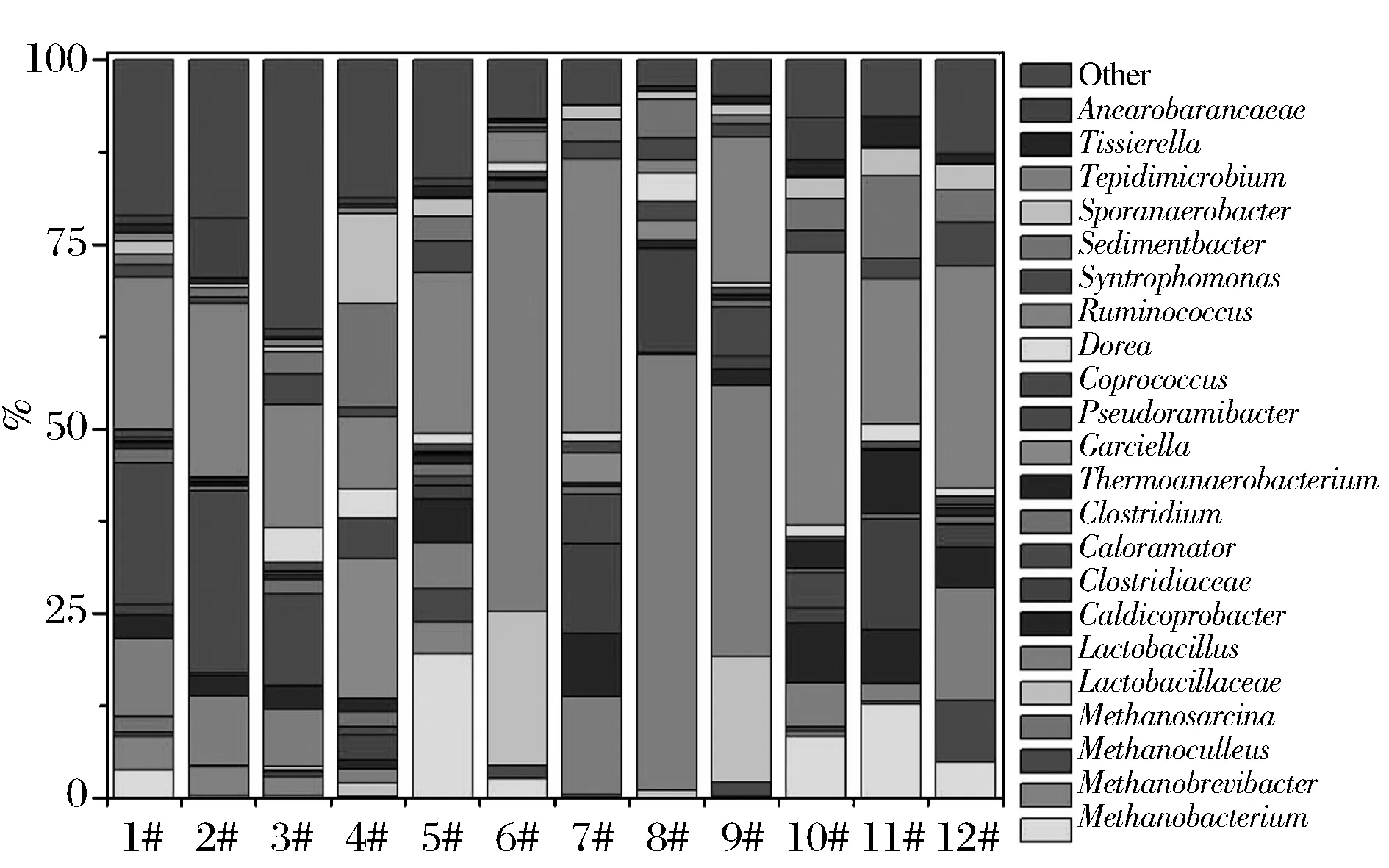
图3 科属水平优势原核微生物群落Fig.3 Dominant prokaryotic microbial community at genus levels in pit muds

图4 五粮液窖泥优势OTU序列系统进化树Fig.4 Phylogenetic tree of the dominant OTUs sequences in pit muds采用邻接法对丰度大于1%的OTUs构建系统发育树。分支点的数字为分子的Bootstrap值(%)。标尺表示0.05核酸距离,分支长度为相似度。树枝末端信息包含分类的种名、OTUs编号和NCBI登录号。
2.2.3 优势序列系统进化分析
上述结果得到了五粮液不同窖泥中总共有17个OTU。RDP Classifier、NCBI Blastn和系统进化分析进一步确认了这17个OTU在属水平上的分类,优势OTU的16S rDNA序列系统发育树如图4所示。它们与其最近的参考序列相似性分析可知,OTU975918与Lactobacillusacetotolerans相似性100%、OTU742140与Garciellasp.相似性90%、OTU4312474与Soehngeniasp.相似性100%、OTU4320991与unclturedTissierellasp.相似度100%、OTU4390692与Sytrophomonas属相似度100%、OTU4411374与unclturedCaloribacteriumsp.相似度100%、OTU771083与unculturedClostridumsp.相似性100%、OTU349343与Eubacteriumcontortum相似度99%、OTU264516与Methanosarcina属相似度100%、OTU283331与Methanoculles属相似度100%、OTU105600与Mthanobacterium属相似度100%,根据相似性分析结果可知,以上OTU的归属基本可以确定,即OTU4411374从QIIME检索结果进一步确认为Caloribacterium、OTU585881由Coprococcus→Clostridium、OTU349343由Dorea→Eubacterium、OTU771083由Rummincoccus→Clostridium、OTU4312474由Sporanaerobacter→Soehngenia。
2.2.4 不同窖泥原核微生物群落相似性分析
将不同样品中属于相同物种的reads数据,利用weighted unifrace算法计算样本间距离矩阵,绘制PCoA主坐标分析图(图5a)。可知,样品6#、8#、9#与其他样品的主要区别在PC1轴上显著区分。基于优势科属原核微生物的丰度(图3)构建样品间关系的主成分分析结果如图5b所示,与PCoA分析结果类似的,样品6#、8#、9#位于PC1轴的正半轴,与6#、8#、9#显著相关的为Lactobacillus和Lactobacillacea。而其余窖泥样品均位于PC1的负半轴,4#、5#和11#位于PC2轴的正半轴,与之显著正相关的包括Sedimentbacter、Sporanaerobacterium和Garciella;1#、2#、3#、7#、10#、12#等样品位于PC2轴的负半轴,与之显著正相关的包括Caloramator、Ruminococcus、Pseduoramibacter等。
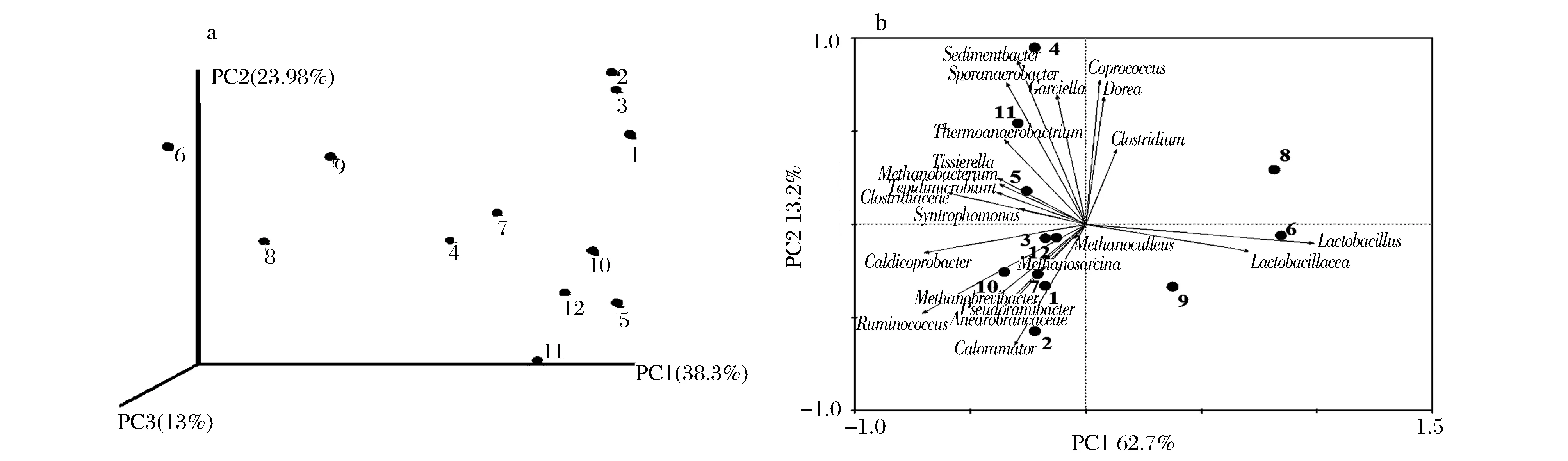
图5 各窖泥样品的Weighted unifrace PCoA图(a)和PCA图(b)Fig.5 Weighted unifrace PCoA analysis (a) and PCA analysis of pit muds (b)
3 讨论
本研究中单个样品检出了超过40万条有效DNA序列。过去文献报道中,利用Roche 454测序仪获得窖泥微生物16S rDNA总有效序列范围为6 568-10 992条[11]。基于Illumina Miseq平台分析浓香型窖泥的总有效序列范围为12 556-65 500条[13,16]。可见,采用本研究所述方法体系获得的测序深度远远大于以上2个平台,故该测序覆盖度具有明显的优势(表1)。
厚壁菌门细菌是五粮液窖泥中的绝对优势菌群(图2),其他同香型窖泥中也检出了类似的结果。利用MiSeq分析四川蜀之源和丰谷不同窖龄窖泥细菌群落中检出了8个细菌门和2个古菌门,其中变形菌门和厚壁菌门是绝对优势菌群[13]。利用Roche焦磷酸测序从丰谷不同窖龄窖泥中检出19个细菌门,其中厚壁菌门占79%[12]。利用焦磷酸测序从四川某知名浓香型酒厂不同窖龄窖泥中检出19个细菌门和2个古菌门,其中厚壁菌门占66.8%[17]。由此可见,同香型(即浓香型)窖泥原核微生物群落组成大类上具有一定的相似性。
但是,五粮液窖泥中优势细菌科属的种类、丰度与其他窖泥的差异显著。本研究检出了大量细菌属,尤其是厚壁菌门(Firmicutes),共检出74个属,如Clostridium,Bacillus,Lactobacillus,Coprococcus、Caldicoprobacter,Soehngenia,Eubacterium,Planococcus,Solibacillus,Macrococcus,Ruminococcus,Sedimentibacter,Thermoanaerobacterium,Caldicoprobacter等,而在其他酒厂窖泥中厚壁菌门细菌则少于本研究,其检出了32个属[11]。同时,两者间优势菌群的构成以及在总菌群中的占比差异显著,本研究中优势厚壁菌门细菌属较多,包括Lactobacillus,Caldicoprobacter,Caloramater,Clostridium,Caloribacterium,Garciella,Eubacterium,Syntrophomonas,Sedimentbacter,Sporanaerobacter,Tissierella(图3),而后者中的优势厚壁菌门细菌属则为Lactobacillus(4.23%~62.28%),Bacillus(0.03%~3.63%),Clostridium(0.03%~3.63%),Sedimentibacter(1.57%~4.78%)和Sytrophomonas(0.45%~4.74%)。
以往的研究中发现酸类、酯类及醇醛类是名优浓香型白酒独特风味的骨架香味成分,酯类主要以乙酯为主(己酸乙酯、乳酸乙酯和乙酸乙酯);同时,优质酒中总酸、总酯的含量较普通白酒高数倍,名优白酒间风格的区别在于三大酯类的含量与相互间的比例不同;酸类是酯类产生的基础,酸多酯高[23]。研究结果发现,Lactobacillus是产生乳酸的主要细菌菌群,Clostridium中Cl.kluyveri是产生己酸、丁酸和H2的主要种属[18]。本研究中,五粮液窖泥中Clostridium(22%)是优势厚壁菌门细菌。Caldicoprobacter(4.1%)属厌氧菌,C.oshimai,C.guelmensis均能利用葡萄糖、乳糖等产生乙酸、醋酸、CO2和H2[19]。毛罗菌科Coprococcus(1.5%)细菌属严格厌氧细菌,C.catus能进行丙酸发酵[20]。Soehngenia(2.7%)细菌中S.saccharolyticagennov.,sp. nov.能代谢酵母膏、半胱氨酸等产生苯乙醛[21]。
甲烷菌常与己酸菌共栖互营生长,并通过种间“氢转移”作用能有效提高浓香型白酒己酸乙酯等的含量[22]。本研究中检出了4个优势的古菌属,其中Methanobacterium、Methanobrevibacter和Methanoculleus属氢营养型甲烷菌,Methanosarcina可同时利用乙酸和H2。四川某知名浓香型白酒窖泥中优势古菌为Methanosarcina,Methanobacterium,Methanobrevibacter和Methanoculleus[17]。本研究中这些甲烷菌的丰度均高于其他窖泥中的丰度,表明甲烷菌群促进己酸乙酯合成的代谢能力特征较为特殊。由于五粮液独特的酿酒工艺,如五种粮食配方和双轮底发酵工艺,该工艺使得下层糟醅与窖泥间相互接触的时间较普通糟醅发酵工艺的时间长1倍甚至更长时间,微生物以及营养物质在两轮甚至三轮发酵的过程中在糟醅与窖泥相接处的界面相互交换,窖泥吸收糟醅中的营养物质,再在微生物尤其是产酸细菌的作用下代谢生成较高含量的酸类等,而这些香气物质又在窖池内发酵水蒸气的作用下转移至糟醅中,糟醅中酵母菌、霉菌等产生的酯化酶类促进酸醇酯化,此外,窖泥本身含有的窖香物质在发酵过程中转移至糟醅,此工艺大大提高了白酒中香气物质的含量以及增加酒体的窖香味。
本研究中,不同空间位置窖泥中原核微生物群落的组成差异明显(图3)且与之关联的原核微生物具有显著差别(图5b),这是由于窖池内糟醅中水分(黄水)从上层糟醅往底层糟醅缓慢浸落,处于底层的窖泥长期处于含水量极高的下层糟醅接触,糟醅中的部分微生物随黄水的滴落而迁移至窖池底部,进而下层窖泥中原核微生物群落的结构与中上层既有一定相似性,又有显著差异。同时本研究发现有部分窖池中层窖泥的泉古菌门丰度高于下层窖泥,而部分窖池则呈相反情况(图3),可能的原因有多种,原因之一可能是由于窖龄的差异,窖池内长期厌氧、高酸等环境造成了窖龄较大的样品中甲烷菌丰度高于中层窖泥(图2);原因之二可能是长期生产过程中班组根据每轮次糟醅状况进行“看糟配料”工艺的差异,即根据每轮次糟醅的实际状况控制五粮粉、曲粉和水的用量。鉴于本研究中样品采集量较少,大量样品采集并归纳总结其中的空间位置规律将是未来的研究重点。
综上所述,本研究通过高通量测序手段调查了五粮液不同类型窖泥的原核微生物群落结构与多样性,发现由于酿酒环境及工艺的独特性,五粮液窖泥中蕴藏了显著区别于其他浓香型白酒窖泥的原核微生物群落,尤其是群系复杂的厚壁菌门细菌、较高丰度的产己酸菌群以及促进己酸生成的甲烷菌群。因此,弄清五粮液酿酒生态各环节中微生物群落多样性以及在传统工艺操作条件下的微生物群落生理代谢特征、演替规律以及与环境的相关性等将是我们下一步重点研究的内容。
[1] 吴衍庸,薛堂荣,陈昭荣,等.五粮液老窖厌氧菌群的分布及其作用的研究[J].微生物学报,1991,31(4):299-307.
[2] 赵东,乔宗伟,彭志云.浓香型白酒发酵过程中酒醅微生物区系及其生态因子演变研究[J].酿酒科技,2007(7):37-39.
[3] AMANN R I,LUDWIG W,SCHLEIFER K H.Phylogenetic identification and in-situ detection of individual microbial-cells without cultivation[J].Microbiology Reviews,1995,59(1):143-169.
[4] 陶勇,徐占成,李东迅,等.窖泥细菌群落结构演替及其与环境因子的相关性[J].酿酒科技,2011(9):42-46.
[5] ZHENG J,LIANG R,ZHANG L Q,et al.Characterization of microbial communities in strong aromatic liquor fermentation pit muds of different ages assessed by combined DGGE and PLFA analyses[J].Food Research International,2013,54(1):660-666.
[6] ZHENG J,WU C,HUANG J,et al.Spatial distribution of bacterial communities and related biochemical properties in Luzhou-flavor liquor fermented grains[J].Journal of Food Science,2014,79(12):M2 491-M2 498.
[7] 郑佳,张良,沈才洪,等.浓香型白酒窖池微生物群落特征[J].应用生态学报,2011,22(4):1 020-1 026.
[8] 刘琨毅,卢中明,郑佳,等.浓香型白酒窖泥微生物群落PLFA指纹图谱方法[J].应用与环境生物学报,2012,18(5):831-837.
[9] 秦臻,郑佳,彭昱雯,等.生物标记法剖析传统酿造用大曲微生物群落结构[J].食品科学,2011,32(11):165-170.
[10] 秦楠,栗东芳,杨瑞馥.高通量测序技术及其在微生物学研究中的应用[J].微生物学报,2011,54(4):445-457.
[11] 陶勇,芮俊鹏,李家宝,等.浓香型白酒窖泥中细菌和古菌的组成与多样性[J].化工学报,2014,65(5):1 800-1 807.
[12] 邓杰,黄治国,卫春会,等.基于高通量测序的浓香型白酒窖池细菌群落结构分析[J].现代食品科技,2015,31(7):50-55.
[13] 李克亚,文章,邓斌,等.不同窖龄窖泥原核生物多样性的高通量测序研究[J].食品工业,2016,36(6):121-125.
[14] Sequencing platform comparison tool[OL].http://www.illumina.com.cn/systems/sequencing-platform-comparison.html.
[15] 温崇庆,何瑶瑶,薛明,等.高通量测序分析DNA提取引起的对虾肠道菌群结构偏差[J].微生物学报,2016,56(1):130-142.
[16] 于春涛,项明,王蕾.浓香型白酒窖泥变质前后古菌群落差异分析[J].酿酒科技,2016(8):60-64.
[17] TAO Y,LI J,RUI J,XU Z,et al.Prokaryotic communities in pit mud from different-aged cellars used for the production of Chinese Strong-flavored liquor[J].Applied and Environmental Microbioliogy,2014,80(7):2 254-2 260.
[18] SEEDORF H,FRICKE W F,VEITH B,et al.The genome ofClostridiumkluyveri,a strict anaerobe with unique metabolic features[J].PNAS,2008,105(6):2 128-2 133.
[19] BOUANANE-DARENFED A,HANIA W B,HACENE H,et al.Caldicoprobacterguelmensissp.nov.a new thermophilic anaerobic,xylanolytic bacterium isolated from an Algerian hot spring[J].International Journal of Systematic Evolution and Microbiology,2013,63:2 049-2 053.
[20] REICHARD N,DUNCAN S H,YOUNG P,et al.Phylogenetic distribution of three pathways for propionate production within the human gut microbiota[J].ISME Journal,2014,8:1 323-1 335.
[21] PARSHINA S N,KLEEREBEZEM R,SANZ J L,et al.Soehngeniasaccharolyticagen.nov.,sp.nov.andClostridiumamygdalinumsp.nov.,two novel anaerobic,benzaldehyde-converting bacteria[J].International Journal of Systematic Evolution and Microbioliogy,2003,53(6):1 791-1 799.
[22] 吴衍庸,刘光烨.窖泥甲烷细菌与浓香型曲酒[J].酿酒,1988(2):29-32.
[23] 王忠彦,尹昌树.白酒色谱骨架成分的含量及比例关系对香型和质量的影响[J].酿酒科技,2000(6):93-96.
ProkaryoticmicrobialcommunityinWuliangyepitmudusingNextSeqhigh-throughputsequencingtechnology
ZHAO Dong,ZHENG Jia*,PENG Zhi-yun,LYU Xue-lan, YANG Kang-zhuo,ZHANG Jian-min
(Technology Research Center of Wuliangye Yibin Co. Ltd.,Yibin 644000,China)
In order to well-understand the specific brewing microbial community of Wuliangye pit mud and its function. The 16S ribosomal DNA V4 sequences of prokaryotic community in Wuliangye pit muds from different positions were investigated by Illumina NextSeq 500 next-generation sequencing (NGS) platform for the first time. Numbers of DNA sequences and operation taxonomy units (OTUs) were summarized by optimized QIIME program, and the abundance and diversity indices of prokaryotic microbial community were calculated. A total of 7 555 164 high quality sequences were obtained and under the same sequencing depth, bacteria (92.3%) and archea (7.6%) were determined as main microbial communities. 22 phyla, 52 classes, 89 orders, 168 family, 330 genera were identified by optimized alignment method. Fimicutes was the predominant bacteria in all pit muds with the abundance of 85.6%,Clostridium,Lactobacillus,Coprococcus,Caldicoprobacter, andSoehngeniawere dominant genus. Furthermore, according to the phylogenetic analysis of dominant OTUs (>1%), the dominant genus in pit mud includedLactobacillus,Caldicoprobacter,Caloramater,Clostridium,Caloribacterium,Garciella,Eubacterium,Syntrophomonas,Sedimentbacter,Sporanaerobacter,Tissierella,Methanosarcina,Methanobacterium,Methanobrevibacter, andMethanoculleus. Wuliangye pit mud contained the specific prokaryotic community in comparison with otherStrong-aromaliquors, and it consisted of complex Firmicutes, high percentage of hexanoic acid-production community and methane community. The specific prokaryotic community can promote the formation of hexanoic acid.
Wuliangye; pit mud; prokaryotic community; NGS
教授级高工,硕士生导师,中国酿酒大师(郑佳研究员为通讯作者,E-mail:zhengwanqi86@163.com)。
固态发酵资源利用四川省重点实验室开放基金(2015GTY005);宜宾市重点科技项目(2016GY009)
2017-02-26,改回日期:2017-05-12
10.13995/j.cnki.11-1802/ts.014156
