AgI/h-MoO3异质结的构筑及其模拟燃油光催化氧化脱硫活性
2017-11-01甄延忠付梦溪张应铮
甄延忠 王 杰 付梦溪,2 张应铮 付 峰
AgI/h-MoO3异质结的构筑及其模拟燃油光催化氧化脱硫活性
甄延忠*,1王 杰1付梦溪1,2张应铮1付 峰*,1
(1陕西省化学反应工程重点实验室,延安大学石油工程与环境工程学院,延安 716000)
(2西安工业大学材料与化工学院,西安 710072)
利用沉积法获得了异质结AgI/h-MoO3光催化剂,通过X射线衍射(XRD)、扫描电子显微镜(SEM)、光电子能谱(EDS)、X射线光电子能谱(XPS)、紫外-可见漫反射吸收光谱(UV-Vis-DRS)、光致发光(PL)、电化学阻抗(EIS)等方法对其物相组成、形貌、光吸收特性、光电化学性能等进行了表征。以噻吩的正辛烷溶液模拟催化裂化(FCC)汽油为探针考察了AgI/h-MoO3光催化氧化脱硫活性,结果表明,AgI/h-MoO3-18异质结在催化剂浓度为1.5 g·L-1,可见光照射2 h后,光催化氧化脱硫活性达98%。利用XRD、XPS、UV-Vis-DRS揭示了AgI/h-MoO3光照后生成少量的金属Ag,使其结构转变为Z型AgI/Ag/h-MoO3,有利于光生电子(e-)转移。利用活性物种捕获实验、循环实验研究了AgI/h-MoO3光催化氧化脱硫机理及其稳定性,实验结果表明:AgI/h-MoO3不仅具有较高的光催化氧化脱硫活性,而且还有良好的稳定性。
沉积法;AgI/h-MoO3异质结;可见光;光催化氧化脱硫
0 引 言
近年来,光催化技术因具有催化活性高、成本低、环境友好等特点引起了人们广泛关注[1-3]。TiO2基光催化剂禁带宽(3.2 eV),光生电子(e-)和空穴(h+)易复合,使其具有较低的太阳光利用率和量子效率,限制了其在光催化领域的实际应用[4]。因此,开发新型光催化剂,提高太阳光利用率和量子效率,仍是当前光催化领域的研究热点[5-6]。
h-MoO3具有独特的一维孔道结构,因而展现了良好的光、电性能[7-8]。2013年,Bose课题组首次报道多级花状结构的h-MoO3具有较窄的禁带宽度(2.94 eV),可作为一种可见光响应的光催化剂[9]。同年,该课题组将h-MoO3与α-MoO3的光催化活性进行了对比,发现h-MoO3在可见光区降解亚甲基蓝(MB)活性较高,说明了h-MoO3是一种性能优异的可见光响应的催化剂[10]。 2015 年,Bose 课题组用 HNO3、HCl对h-MoO3带隙进行了调节,研究发现,h-MoO3(HNO3)、h-MoO3(HCl)的带隙值分别为 2.94、3.04 eV,光催化实验表明h-MoO3(HCl)在可见光区降解MB活性较高[11]。另外,曹丽云教授课题组考察了前驱体中Mo浓度对h-MoO3光催化性能影响,研究表明:前驱体中 Mo浓度为 0.07 mol·L-1时制备的 h-MoO3微晶形貌规则,对亚甲基蓝具有较好的光催化活性[12]。
AgX(X=Cl、Br、I)作为可见光响应的光催化剂,得到了人们广泛研究[13]。然而,AgX不稳定,可见光照射后易分解,阻碍了AgX在光催化领域的实际应用,因此人们经常利用沉积法将AgX负载到稳定半导体表面,构成异质结光催化剂,从而提高其稳定性和光催化活性[14]。 近年来,AgI/TiO2[15-16]、AgI/BiPO4[17]、Ag3PO4/AgI[18]、Bi2SiO5/AgI[19]、AgI/BiVO4[20]等可见光催化剂被不同课题组报道。近期研究发现,AgX异质结在光催化反应初期表面生成少量的金属Ag有利于光生e-/h+的分离,不仅可提高光催化活性而且还能提高其稳定性。例如,2013年,Chen等发现Ag3PO4/AgI表面生成少量的金属Ag,使其转变为Z型Ag3PO4/Ag/AgI,该催化剂在可见光照射18 min后,对甲基橙(MO)、苯酚(RhOH)的降解率分别为96.9%、90.0%,循环使用5次后对MO仍具有较高的光催化活性[18]。2016年,Yang等发现AgI/BiVO4在光催化降解四环素 (TC)初期形成Z型AgI/Ag/BiVO4结构,在可见光照射120 min后降解率可达94.91%,循环使用4次后降解率仍为94.91%[20]。
另外,通过对AgI、h-MoO3带隙结构计算,AgI导带(CB)电势(-0.50 eV)比 h-MoO3导带电势(0.39 eV)更负,而 h-MoO3价带(VB)电势(3.41 eV)比 AgI价带电势(2.31 eV)更正,因此二者的能带结构匹配,形成的异质结光催化剂有利于光生载流子的分离,使h-MoO3光催化活性提高。基于此,本文利用沉积法制备了一种新型的异质结光催化剂,AgI/h-MoO3,通过 XRD、SEM、EDS、XPS、UV-Vis-DRS、PL、EIS 等方法对其组成、形貌、光吸收性能、光电化学性能等进行了表征。选用噻吩的正辛烷溶液模拟催化裂化(FCC)汽油,研究了AgI/h-MoO3光催化氧化脱硫活性。利用XRD、XPS、UV-Vis-DRS分析,发现光照后AgI/h-MoO3中生成了少量金属Ag,同时结合活性物种捕获实验和循环实验详细研究了AgI/h-MoO3光催化氧化脱硫机理及其稳定性。
1 实验部分
1.1试 剂
(NH4)6Mo7O24·4H2O、HNO3(含量 6l%~63%)购于北京化工厂,AgNO3、KI、噻吩、正辛烷、无水乙醇(CHCH2OH)购于天津凯通化学试剂有限公司,所有试剂均为AR分析纯。全部实验用水均为去离子水。
1.2 催化剂的制备
h-MoO3的制备:称量 1.236 g(NH4)6MO7O24·4H2O溶于10 mL去离子水中,磁力搅拌30 min,形成均一溶液后,加入10 mL浓度为2 mol·L-1HNO3溶液,继续搅拌30 min,最后将混合溶液移入聚四氟乙烯不锈钢高压反应釜,放入电热恒温鼓风干燥箱中升温到140℃下持续12 h。反应结束后,自然冷却至室温,依次经离心、水洗、醇洗数次,80℃下干燥12 h得到白色样品。
AgI/h-MoO3异质结的制备:量取去离子水30 mL 4份,分别加入质量为0.144 g的h-MoO3,超声20 min 后, 分别逐滴加入 2.7、6.3、11.3、18.5 mL 的AgNO3溶液(0.1 mol·L-1),黑暗环境下磁力搅拌 1 h后再超声 20 min,随后分别将 2.7、6.3、11.3、18.5 mL浓度为0.1 mol·L-1的KI溶液逐滴加入上述溶液中,继续搅拌10 h,高速离心分离所得沉淀,依次用去离子水、无水乙醇各洗3次,80℃下真空干燥12 h, 得到负载量分别为 20%、38%、52%、64%(w/w)的AgI/h-MoO3样品,分别被标记为AgI/h-MoO3-6、AgI/h-MoO3-12、AgI/h-MoO3-18、AgI/h-MoO3-24。
1.3 催化剂的表征
样品的物相组成在日本岛津公司XRD-7000型全自动X射线粉末衍射仪上测定,Cu Kα(Ni滤玻片,λ=0.154 18 nm),管电压 40 kV,管电流 30 mA;样品的光电子能谱在美国PE公司PH5400型光电子能谱仪上检测,铝靶Al Kα,电压1 486.68 eV,激发功率250 W,用C1s电子结合能284.6 eV进行误差校正;样品的形貌在日本电子JEOL-6701型场发射扫描电子显微镜 (FE-SEM)上观察,工作电压5 kV;样品紫外-可见吸收光谱在日本岛津UV-2550型紫外-可见分光光度计测定,标准BaSO4作参比。光致发光(PL)图谱测定在美国Spex 500M荧光光谱仪上进行,激发波长为225 nm。电化学阻抗(EIS)测定在CHI660E电化学工作站上进行,以铂电极、饱和甘汞电极和样品分别作为对电极、参比电极和工作电极的三电极系统进行分析,用0.1 mol·L-1的Na2SO4溶液作为电解液。
1.4 光催化氧化模拟FCC汽油脱硫实验
取0.5 mL噻吩加入500 mL正辛烷溶液中充分混匀配制成模拟催化裂化汽油(FCC汽油),其硫含量约为500 mg·L-1。量取20 mL上述溶液和称取一定量的催化剂加入到50 mL石英试管中,为了检测生成产物,在另一只试管中再加入5 mL Ba(OH)2稀溶液,试管下部为水相上部为油相。将石英试管在黑暗中磁力搅拌30 min,达到吸附-脱附平衡。然后打开350 W的氙灯照射,同时打开旋转开关,保证受光均匀。每隔30 min取样,然后用乙腈进行萃取,取其上清液在微库伦分析仪中分析其硫含量的大小。含硫量的测定在WK-2D型库仑分析仪上进行,汽化室温度为680℃,燃烧室温度为850℃,以氮气作为载气,氧气为燃烧气,碘电极为参比电极,铂电极为测量电极。按下式计算脱硫率,其中η为脱硫率,C0为原液中硫含量;C1为反应后溶液中硫含量。

2 结果与讨论
2.1 XRD和XPS分析
通过XRD对h-MoO3、AgI与不同负载量的AgI/h-MoO3异质结的晶相、组成进行了表征,结果如图1所示。由图可知,h-MoO3衍射峰的位置、d值与标准卡(PDF No.21-0569)一致,说明所得样品为六方晶系的h-MoO3,图中无杂峰且衍射峰的强度较高,说明了样品纯度、结晶度较高[9]。AgI各衍射峰的位置、d值与标准卡(PDF No.09-0399)相吻合,2θ在 23.79°、39.26°、46.31°处的衍射峰对应的(111)、(220)、(311)晶面,因此所得样品为立方面心晶系的γ-AgI[16]。在AgI/h-MoO3异质结的 XRD图中只有 γ-AgI和 h-MoO3的特征峰,无其它杂质峰,说明没有新物相生成,同时,γ-AgI衍射峰的强度随着负载量的增加而增强。

图 1 h-MoO3、AgI与 AgI/h-MoO3的 XRD 图Fig.1 XRD patterns of h-MoO3,AgI and AgI/h-MoO3
利用XPS对AgI/h-MoO3-18的表面元素组成、价态进行了分析,结果如图2所示。由图2(a)可知,样品中含有 Mo、O、Ag、I、C 元素, 其中 C 元素来源于仪器。图2(b)为Mo的高分辨图谱,由图可知,结合能 232.6、235.8 eV 处对应 Mo3d5/2、Mo3d3/2特征峰,表明样品中Mo6+[21]。O1s经拟合得到3个峰(如图2(c)所示),结合能 530.4 eV处归属为 Mo-O,531.5 eV处为Mo-OH,533.3 eV处则归属为样品表面H2O中 O1s的特征峰[21-23]。 图 2(d,e)分别为 Ag、I的高分辨图谱,Ag3d5/2、Ag3d3/2特征峰的结合能分别为367.9、373.9 eV,说明了样品中存在 Ag+;I3d5/2、I3d3/2特征吸收峰的结合能为619.4、630.9 eV,说明了样品中存在I-,实验结果与文献报道一致[24]。
2.2 SEM分析
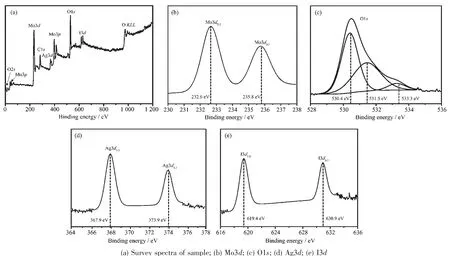
图2 AgI/h-MoO3-18的XPS图谱Fig.2 XPSspectra of AgI/h-MoO3-18

图3 h-MoO3与AgI/h-MoO3的SEM照片Fig.3 SEM images of h-MoO3 and AgI/h-MoO3
图 3为 h-MoO3、AgI/h-MoO3异质结的 SEM 照片。由图3(a)可知,所得h-MoO3为六棱柱形状,直径大约为5μm,棱柱长约为几十微米,表面光滑,无团聚现象。图3(a)中的插图为h-MoO3的EDS图,由此可知,样品仅由Mo、O两种元素组成,原子个数比为1∶3,表明所得样品为纯 h-MoO3。 图 3(b,e)分别是异质 结 AgI/h-MoO3-6,AgI/h-MoO3-12,AgI/h-MoO3-18,AgI/h-MoO3-24的SEM照片,由图可知,样品中存在2种相,深色为h-MoO3,白色亮点为AgI,其原因是AgI具有较高的离子电导率[25]。图3(d)的插图为AgI/h-MoO3-18样品的EDS图,由图可知,样品由Mo、O、Ag、I组成,其中 Ag、I的原子个数比为 1∶1,表明了h-MoO3表面负载的为AgI。负载后h-MoO3形状保持不变,但是表面变粗糙。同时,随着AgI负载量的增加,h-MoO3表面AgI的粒径不断增大,当负载量为64%时,出现了团聚现象(如图2(e)所示)。
2.3 紫外-可见漫反射吸收光谱、光致发光、电化学阻抗
图 4(a)为 h-MoO3、AgI与 AgI/h-MoO3异质结的紫外-可见漫反射吸收(UV-Vis-DRS)光谱,由图可知,h-MoO3、AgI在可见光区均有吸收,h-MoO3吸收边界为431 nm,AgI的吸收边界为458 nm。与之相比,AgI/h-MoO3的吸收边界发生明显的红移,AgI/h-MoO3-6、AgI/h-MoO3-12、AgI/h-MoO3-18、AgI/h-MoO3-24 的吸收边界大约在 472、473、474、475 nm。
半导体的带隙与带边吸收的关系为:αhν=(hν-Eg)n,其中,α、h、ν和 Eg分别是样品的吸收系数、普朗克常数、光子频率和带隙值。h-MoO3的n值为2,作(αhν)1/2-hν图,计算带隙值[9,26],结果如图 4(b)所示。 h-MoO3、AgI、AgI/h-MoO3-6、AgI/h-MoO3-12、AgI/h-MoO3-18与 AgI/h-MoO3-24带隙值分别为 3.02、2.81、2.78、2.75、2.73、2.72 eV, 由此可知,AgI与 h-MoO3的复合降低了催化剂的带隙值,提高对可见光的吸收能力。

图 4 (a)h-MoO3、AgI与 AgI/h-MoO3的紫外-可见漫反射吸收光谱,(b)(αhν)1/2-hν曲线Fig.4(a)UV-Vis-DRSspectra of h-MoO3,AgIand AgI/h-MoO3 with different loading amounts,(b)Plots of the(αhν)1/2 vs hν

图5 h-MoO3、AgI/h-MoO3-18光致发光图谱Fig.5 PL spectra of h-MoO3,AgI/h-MoO3-18
利用光致发光(PL)谱揭示催化剂光生e-/h+的相对寿命。一般情况下,PL峰的强度越低,表明光生e-、h+的复合率较低,光生载流子寿命较长[27]。由图5可知,h-MoO3有3个特征发射峰,其中在410 nm处的发射峰是自由激子之间的激子-激子碰撞发光,而460、545 nm处的特征峰则是光生 e-、h+复合发光,实验结果与文献报道一致[9]。AgI/h-MoO3-18的PL发光强度明显低于h-MoO3,在460、545 nm处更为显著,说明了AgI对h-MoO3表面的修饰,有效的抑制光生e-、h+的复合,延长光生载流子的寿命。另外,AgI/h-MoO3-18在410 nm处的发射峰有轻微的红移,这是由于AgI的修饰改变h-MoO3表面的电荷密度分布[28-29]。
文献中常用EIS研究催化剂光生载流子的迁移和转移过程,在EIS图中圆弧半径越小说明界面层电阻越小,反应速率越快,光生e+/h+复合速率越低[30-31]。图 6为 h-MoO3、AgI与 AgI/h-MoO3-18的 EIS图谱,由图可知,AgI/h-MoO3-18的圆弧半径比h-MoO3、AgI的小,说明了AgI/h-MoO3-18电荷转移效率更高,AgI的负载有助于提高光生e+/h+对的分离效率,从而提高光催化活性。
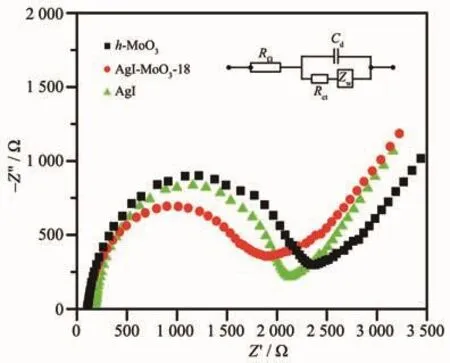
图 6 h-MoO3、AgI、AgI/h-MoO3-18 电化学阻抗图谱Fig.6 EISpatterns of h-MoO3,AgI and AgI/h-MoO3-18
2.4 光催化脱硫活性
以噻吩的正辛烷溶液模拟FCC汽油,以空气为氧化剂,氙灯模拟可见光,考察了催化剂的光催化氧化脱硫性能,结果如图7所示。图7(a)为h-MoO3、AgI和AgI/h-MoO3光催化氧化脱硫率随时间的变化曲线,由图可知,AgI/h-MoO3的光催化活性高于h-MoO3、AgI,且随着负载量的增加而提高。可见光照射 2h 后,h-MoO3、AgI、AgI/h-MoO3-6、AgI/h-MoO3-12、AgI/h-MoO3-18、AgI/h-MoO3-24脱硫率分别为17%、74%、82%、85%、98%、88%。AgI/h-MoO3-24脱硫率低于AgI/h-MoO3-18,其原因为AgI在h-MoO3表面发生了团聚现象,影响了光生e-/h+分离,导致光催化活性下降[32]。在混有Ca(OH)2溶液的试管中,试管壁上产生大量的白色沉淀,取其少量加入HNO3,沉淀部分溶解,说明产物中含有SO3和CO2,实验现象与文献报道一致[33-34]。
噻吩的光催化氧化降解满足一级动力学方程,其中C0′为暗反应结束后的硫含量,Ct为光催化降解过程中不同时间段的硫含量,k为表观速率常数[35]。

由图 7(b)可知,AgI/h-MoO3-24、AgI/h-MoO3-18、AgI/h-MoO3-12、AgI/h-MoO3-6、AgI与 h-MoO3的表观速率常数分别为 0.019 9、0.026 0、0.015 3、0.013 2、0.010 0、0.001 9 min-1。 其中,AgI/h-MoO3-18 分别是h-MoO3、AgI的 13.7 倍、2.6 倍。

图7 h-MoO3、AgI和AgI/h-MoO3光催化性能:(a)不同催化剂的脱硫率,(b)光催化氧化脱硫一级反应动力学图谱,(c)催化剂浓度对光催化活性的影响,(d)催化剂循环实验Fig.7 Photocatalytic performances of h-MoO3,AgI,and AgI/h-MoO3:(a)desulfurization efficiency of difference catalyst,(b)first-order kinetic fit for photodegradation for thiophene,(c)Effects of catalyst dosage on photocatalytic activity,(d)cycling run of the sample
另外,考察了催化剂浓度与光催化活性之间的关系,结果如图7(c)所示。由图可知,当AgI/h-MoO3-18 浓度从 0.5 g·L-1增加至 1.5 g·L-1,脱硫率从 77%增加至98%,当浓度增至2.0 g·L-1时,脱硫率下降至92%。其原因为:适量的催化剂不仅提高光的利用率,而且增加了反应的活性位点,所以光催化活性提高;催化剂浓度过大则会发生团聚,使反应中的活性位点减少,所以光催化活性降低[36]。
图7(d)为AgI/h-MoO3-18循环实验,由图可知,重复循环使用5次后,催化剂的光催化氧化脱硫率由98%降低至83%,脱硫活性仍然高于h-MoO3、AgI,说明了AgI/h-MoO3-18具有良好的稳定性。
另外,我们对光照后的AgI/h-MoO3-18进行了XRD、XPS分析,结果如图8所示。由图8(a)可知,光照后AgI/h-MoO3-18的XRD图在38.11°处产生一个非常微弱的衍射峰,可归属为金属Ag(PDF No.04-0783)的(111)晶面。图8(b)为光照后样品中Ag的高分辨XPS图谱,其中374.3、368.8 eV处为金属Ag的 3d5/2和 3d3/2的特征峰,373.9和 368.0 eV处为Ag+的3d3/2和3d5/2的特征峰。由此可知,光照后AgI/h-MoO3中有少量金属Ag生成,其结构转变为Z型AgI/Ag/h-MoO3。根据前期文献报道,金属Ag具有表面等离子体共振效应(SPR)和良好的导电性,利于催化剂光生e-/h+分离,对扩展光吸收范围,提高光催化活性具有重要的作用[20,37]。
图 8(c)为光照前后 AgI/h-MoO3-18的 UV-Vis-DRS性能对比,光照后AgI/h-MoO3-18在可见光区的吸收强度明显增强,吸收边界发生了明显红移,说明了金属Ag的生成有利于扩展AgI/h-MoO3-18的光吸收范围。图8(d)为光照前后的EIS对比图谱,由图可知,光照前后AgI/h-MoO3-18的圆弧半径非常接近。

图8 AgI/h-MoO3-18光照前后对比图:(a)XRD图(插图为局部放大图),(b)Ag3d的XPS图谱,(c)UV-Vis-DRS图,(d)电化学阻抗图谱Fig.8 Comparison of(a)the XRD patterns(the partial amplified pattern in inset),(b)XPSspetrum of Ag3d,(c)UV-Vis-DRSspectrum,(d)EISpatterns for the used and fresh AgI/h-MoO3-18
2.5 光催化氧化脱硫机理
为了获知光催化氧化脱硫过程中的活性物种,选用了乙二胺四乙酸二钠(EDTA-2Na)、异丙醇(IPA)和苯醌 (BQ)分别作为h+、·OH和·O2-自由基的捕获剂[38]。由图9可知,加入EDTA-2Na、BQ时,AgI/h-MoO3-18的光催化活性受到明显抑制,说明在AgI/h-MoO3-18光催化氧化脱硫过程中,h+、·O2-是反应的主要活性物种。IPA的加入对AgI/h-MoO3-18的光催化活性影响不大,说明·OH不是主要的活性物种。

图9 AgI/h-MoO3-18活性物种捕获试验Fig.9 Active species trapping experiments of AgI/h-MoO3-18
催化剂的催化活性与其带隙结构密切相关,利用式 3和 4,计算了 h-MoO3、AgI导带(CB)和价带(VB)位置,数据列于表1。

AgI导带和价带电势分别为-0.50、2.31 eV,h-MoO3的则为0.39、3.41 eV,二者的能带结构匹配,可形成异质结光催化剂,为此按照以下2种方式推测了AgI/h-MoO3光催化氧化脱硫机理。如图10(a)所示,在可见光照射下AgI、h-MoO3均可被激发产生e-/h+,其中 AgI导带电势(-0.50 eV)低于 h-MoO3(0.39 eV),因此AgI导带上的e-容易迁移到h-MoO3导带上。h-MoO3价带电势 (3.41 eV)高于AgI价带(2.31 eV),因此h-MoO3价带的h+很容易迁移到AgI价带。然而,h-MoO3导带电势 (0.39 eV)高于O2/·O2-电势(-0.33 eV vs NHE),所以不能将 O2还原为·O2-,同时AgI价带电势(2.31 eV)低于·OH/OH-电势(2.38 eV vs NHE),因此 h+不能将 OH-氧化为·OH[39],机理 1 与AgI/h-MoO3-18活性物种捕获实验的结果不相符。
光照后,AgI/h-MoO3转变为Z型AgI/Ag/h-MoO3时,光催化氧化脱硫机理如图10(b)所示。在可见光照射下,AgI与h-MoO3均被激发产生了光生e-/h+(式4、5)。金属Ag的费米能级为0.4 eV,高于h-MoO3导带电势(0.39 eV),因此在界面Schottky势垒作用下,h-MoO3导带上的e-非常容易注入金属Ag(式6)[40]。AgI价带电势(2.31 eV)高于金属Ag的费米能级(0.4 eV),因此金属Ag上e-转移到AgI价带并与h+复合,其复合速率快于AgI光生e-/h+的复合(式7)[39]。由此可知,半导体间的e-传输,利于提高AgI/h-MoO3光生e-/h+的分离。在AgI导带(-0.50 eV)e-与O2结合生成了·O2-(式 8),在 h-MoO3价带 h+将 OH-氧化为·OH(式9)。由活性物种捕获实验可知,仅有少量的·OH参与了光催化反应,这与前期文献报道一致[20]。FCC模拟汽油中,硫原子的孤对电子可被h-MoO3价带上h+捕获,形成基态阳离子 C4H4S+(式 10)。 C4H4S+与·O2-结合形成极性极强的氧化噻吩C4H4SO2-(式11),氧化噻吩经过萃取除去,未除去的C4H4SO2-与·O2-结合转化为CO2、SO3和H2O(式12),相关产物在光催化氧化脱硫实验中得到了验证[41]。

表1 半导体的电负性、导带边缘、价带位置和带隙能Table 1 Absolute electronegativity(X),caculated CB edge(E CB),calculated VB(E VB)position and band gap energy(E g)for the semiconductors

图10 AgI/h-MoO3光催化氧化噻吩机理示意图Fig.10 Schematic diagram of mechanism for the photocatalytic oxidation for thiophene by AgI/h-MoO3
具体反应过程如下列方程所示:

3结 论
(1)利用沉积法获得负载量不同的AgI/h-MoO3异质结光催化剂,光催化氧化脱硫实验结果表明,AgI/h-MoO3-18光催化氧化脱硫活性最好,当催化剂浓度为1.5 g·L-1,光照 2 h后,脱硫率高达 98%,循环使用5次后脱硫率仍保持83%以上。
(2)利用 XRD、XPS、UV-Vis-DRS对使用后 AgI/h-MoO3进行分析,发现有少量金属Ag生成,使AgI/h-MoO3结构转变为Z型AgI/Ag/h-MoO3光催化剂。Z型AgI/Ag/h-MoO3有利于光生e-/h+的分离,扩展了光吸收范围,提高了光催化活性。通过活性物种捕获实验探讨了光催化氧化脱硫机理,结果表明了h+、·O2-是AgI/h-MoO3光催化氧化脱硫的重要活性物种。
[1]Fresno F,Portela R,Suárez S,et al.J.Mater.Chem.A,2014,2(9):2863-2884
[2]Hoffmann M R,Choi W,Bahnemann D W.Chem.Rev.,1995,95(1):69-96
[3]FU Rong-Rong(付蓉蓉),LI Yan-Min(李延敏),GAO Shan-Min(高善民),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(10):2231-2245
[4]Zhang F,Zhang CL,Peng H Y,et al.Part.Part.Syst.Char.,2016,33(5):248-253
[5]Wang H L,Zhang L S,Chen Z G,et al.Chem.Soc.Rev.,2014,43(15):5234-5244
[6]Sathish M,Viswanathan B,Viswanath R P,et al.J.Mol.Struct.,2017,318(4):213-217
[7]Pan W,Tian R,Jin H,et al.Chem.Mater.,2010,22(22):6202-6208
[8]Chithambararaj A,Bose A C.Beilstein J.Nanotechnol.,2011,2:585-92
[9]Chithambararaj A,Sanjini N S,Bose A C,et al.Catal.Sci.Technol.,2013,3(5):1405-1414
[10]Chithambararaj A,Sanjini N S,Velmathib S,et al.Phys.Chem.Chem.Phys.,2013,15(35):14761-14769
[11]Chithambararaj A,Winston B,Sanjini N S,et al.J.Nanosci.Nanotechnol.,2015,15(7):4913-4919
[12]MA Feng-Lan(马凤兰),CAO Li-Yun(曹丽云),HUANG Jian-Feng(黄剑锋),et al.J.Chin.Ceram.Soc.(硅酸盐学报),2013,41(6):867-870
[13]ZHANG Su-Yun(张素云),WANG Jia-Xi(汪家喜),WANG Qian(王迁).Chinese J.Inorg.Chem.(无机化学学报),2017,33(7):1181-1186
[14]Shi H,Li G,Sun H,et al.Appl.Catal.,B,2014,158:301-307
[15]Wang Q,Shi X,Xu J,et al.J.Hazard.Mater.,2016,307:213-220
[16]Wang Q,Shi X,Liu E,et al.Ind.Eng.Chem.Res.,2016,55(17):4897-4904
[17]Ye H,Lin H,Cao J,et al.J.Mol.Catal.A:Chem.,2015,397:85-92
[18]Chen Z,Wang W,Zhang Z,et al.J.Phys.Chem.C,2013,117(38):19346-19352
[19]Wan Z,Zhang G.J.Mater.Chem.A,2015,3(32):16737-16745
[20]Chen F,Yang Q,Sun J,et al.ACSAppl.Mater.Interfaces,2016,8(48):32887-32900
[21]Chen Y,Lu C,Xu L,et al.CrystEngComm,2010,12(11):3740-3747
[22]Shakir I,Shahid M,Kang D J.Chem.Commun.,2010,46(24):4324-4326
[23]WANG Dan-Jun(王丹军),SHEN Hui-Dong(申惠东),GUO Li(郭莉),et al.Acta Scieneiae Circumstantiae(环境科学学报),2017,37(5):1751-1762
[24]Cheng H,Huang B,Dai Y,et al.Langmuir,2010,26(9):6618-6624
[25]Ng CH B,Fan W Y.J.Phys.Chem.C,2007,111(26):9166-9171
[26]Sinaim H,Ham D J,Lee JS,et al.J.Alloys Compd.,2012,516:172-178
[27]Li L,Zhang J,Shen C,et al.Fuel,2016,167:9-16
[28]Xiang Q,Yu J,Jaroniec M.J.Phys.Chem.C,2011,115(15):7355-7363
[29]Luo X,Liu P,Truong N T N,et al.J.Phys.Chem.C,2011,115(43):20817-20823
[30]Hosseini Z,Taghavinia N,Sharifi N,et al.J.Phys.Chem.C,2008,112(47):18686-18689
[31]LIANG Yan-Ping(梁 燕 萍 ),JIA Jian-Ping(贾 剑 平 ),LÜ Xiang-Fei(吕向菲),et al.Chinese J.Inorg.Chem.(无机化学学报),2010,26(4):633-638
[32]Shi L,Liang L,Ma J,et al.Catal.Sci.Technol.,2014,4(3):758-765
[33]ZHEN Yan-Zhong(甄延忠),LI Jin(李静),WANG Dan-Jun(王丹军),et al.J.Inorg.Mater.(无机材料学报),2014,30(4):408-412
[34]Li F T,Liu Y,Sun Z M,et al.Catal.Sci.Technol.,2012,2(7):1455-1462
[35]Miao G,Huang D,Ren X,et al.Appl.Catal.,B,2016,192:72-79
[36]ZHAO Di-Shun(赵地顺),LIU Cui-Wei(刘翠微),MA Si-Guo(马四国).Chem.J.Chinese Universities(高等学校化学学报),2006,27(4):692-696
[37]Zhang Y,Tang Z R,Fu X,et al.Appl.Catal.,B,2011,106(3):445-452
[38]Bai X J,Wang L,Zong R L,et al.Langmuir,2013,29(9):3097-3105
[39]Cheng H,Huang B,Dai Y,et al.Langmuir,2010,26(9):6618-6624
[40]Wang Y,Niu C G,Zhang L,et al.RSC Adv.,2016,6(13):10221-10228
[41]Lin F,Li C,Wang D G,et al.Energy Environ.Sci,2012,5(4):6400-6406
AgI/h-MoO3Heterojunctions:Fabrication and Photocatalytic Oxidative Desulfurization Activity of Simulation Fuel
s:AgI/h-MoO3heterojunction photocatalysts were successfully fabricated by a facile deposition method.XRD,SEM,UV-Vis-DRS,EDS,XPS,PL and EISwere employed to characterize the structure,morphology,optical absorption and photo-electrochemical properties of as-synthesized samples.The photocatalytic oxidative desulfurization activity of as-synthesized samples was investigated with thiophene simulated the catalytic cracking(FCC)gasoline as the probe.The results indicate that AgI/h-MoO3-18 heterojunction has the highest photocatalytic activity and itsremoval efficiency for thiophene can be up to 98%with catalyst dosage(1.5 g·L-1)under irradiation of visible light for 2 h.XRD,XPS and UV-Vis-DRS measurements reveal that a small amount of metallic Ag which facilitates photoinduced electrons transfer is formed on the surface of used AgI/h-MoO3,thus leading to the transformation from AgI/h-MoO3to AgI/Ag/h-MoO3.The photocatalytic oxidative desulfurization mechanism and stability performance of AgI/Ag/h-MoO3were also studied by the active species trapping experiment and recycling run experiment.The experimental results have exhibited that the as-prepared AgI/h-MoO3heterojunctions not only have higher photocatalytic oxidative desulfurization activity for thiophene,but also have good stability.
deposition method;AgI/h-MoO3heterojunction;visible light;photocatalytic oxidative desulfurization
O614.61
A
1001-4861(2017)10-1731-10
10.11862/CJIC.2017.236
ZHEN Yan-Zhong*,1WANG Jie1FU Meng-Xi1,2ZHANG Ying-Zheng1FU Feng*,1
(1Shaanxi Key Laboratory of Chemical Reaction Engineering,Department of Petroleum Engineering and Environmental Engineering,Yan′an University,Yan′an,Shaanxi 716000,China)
(2School of Materials Science and Chemical Engineering,Xi′an Technological University,Xi′an 710072,China)
2017-07-17。收修改稿日期:2017-09-10。
国家自然科学基金项目(No.21663030)、陕西科技攻关项目(No.2015GY174)、延安市成果转化项目(No.2016CGZH-12-01)、延安大学科技项目(No.YD2015-13)和创新训练项目(No.YCX201734,1553)资助。
*通信联系人。 E-mail:zyz943@163.com
