双乌止痛酊质量标准研究
2017-11-01钱文慧任海祥
苏 华,钱文慧,廖 欣,白 汝,任海祥
(中国人民解放军南京军区南京总医院制剂科,江苏 南京 210002)
研究
双乌止痛酊质量标准研究
苏 华,钱文慧,廖 欣,白 汝,任海祥
(中国人民解放军南京军区南京总医院制剂科,江苏 南京 210002)
目的制订双乌止痛酊的质量标准。方法采用薄层色谱法对双乌止痛酊中的制天南星、当归及苯甲酰次乌头原碱进行定性鉴别,同时对制剂中的毒性成分乌头碱进行限量检查;采用高效液相色谱法测定苯甲酰新乌头原碱含量,色谱柱选用Inertsustain C18柱(250 mm ×4.6 mm,5 m),以乙腈 -0.02 mol/L 的磷酸二氢钠(25∶75)为流动相,检测波长为 230 nm,柱温为 30℃,流速为 1.0 mL /min。结果双乌止痛酊中制天南星、当归及苯甲酰次乌头原碱的定性鉴别专属性强,乌头碱限量测定符合规定;苯甲酰新乌头原碱的检测质量浓度在 0.012 44~0.099 52 g/L(r=1.000 0,n=5)范围内与峰面积线性关系良好,平均加样回收率为 99.36% ,RSD =2.45%(n=9)。结论该方法操作简单,准确可靠,可用于双乌止痛酊的质量控制。
双乌止痛酊;苯甲酰新乌头原碱;质量标准;薄层色谱法;高效液相色谱法
双乌止痛酊为我院院内制剂,是由制川乌、制草乌、制天南星、细辛、丁香、当归、川芎等十几味中药通过渗漉法制成的外用酊剂,有效成分复杂多样,临床主要用于关节痛、腰腿痛、肩周炎等风湿痹痛的治疗,同时对各种急性扭伤、挫伤等有效,具有祛风散寒、舒筋活络、消炎止痛功效[1]。方中制川乌、制草乌中所含的乌头类生物碱成分是制剂的毒/效成分,两药由生川乌、生草乌炮制而得,炮制后有毒的双酯型生物碱类如乌头碱、新乌头碱、次乌头碱等成分大部分水解转变成苯甲酰乌头原碱、苯甲酰新乌头原碱、苯甲酰次乌头原碱等单酯型乌头碱[2-4],而药效并未发生改变。因此,双乌止痛酊中既含有消炎镇痛的有效成分单酯型乌头碱,又含有毒性的双酯型生物碱(如乌头碱)。现代研究中关于含制乌头类毒/效成分的中成药制剂的质控研究较少。本研究旨在制订双乌止痛酊的质量标准,建立薄层定性鉴别和高效液相色谱定量测定制剂中的多种有效成分,同时结合对主要毒性成分乌头碱的限量测定来全面控制该制剂质量,以期为临床安全使用乌头类成分的制剂提供依据。现报道如下。
1 仪器与试药
1.1 仪器
LC-20A型高效液相色谱仪(日本岛津公司);KQ-250DB型数控超声波清洗器(昆山超声仪器有限公司);FA1604S型电子天平(上海天平仪器厂);AE240型电子分析天平(梅特勒-托利多仪器有限公司)。
1.2 试药
当归对照药材(批号为 120927-201315),川芎对照药材(批号为120918-201110),干姜对照药材(批号为 120942-201309),乌头碱对照品(批号为 110720-201111,纯度为98.8%),苯甲酰次乌头原碱对照品(批号为 111796-201303,纯度为 97.5%),苯甲酰新乌头原碱对照品(批号为111795-201303,纯度为96.3%),均购自中国食品药品检定研究院;薄层层析硅胶G薄层板(青岛海洋化工有限公司);甲醇(色谱醇,美国TEDIA公司);双乌止痛酊(规格为每瓶60 mL,医院自制,批号分别为 160509,160613,160711);其余所用试剂均为分析纯。
2 方法与结果
2.1 薄层色谱鉴别
2.1.1 制川乌、制草乌
对制剂中制川乌、制草乌的有效成分苯甲酰次乌头原碱进行薄层色谱鉴别。取本品10 mL,挥去乙醇,加乙醚20 mL使溶解,加氨试液 2 mL,超声处理 30 min,滤过,滤液蒸干,残渣加二氯甲烷1 mL使溶解,作为供试品溶液。另取苯甲酰次乌头原碱对照品,加异丙醇-三氯甲烷(1∶1)混合溶液制成每1 mL含1 mg的对照品溶液。按处方比例称取除制川乌、制草乌以外的药材,研细,按本品制备工艺及供试品溶液制备方法制成阴性对照品溶液。照薄层色谱法(2010年版《中国药典(一部)》附录Ⅵ B)试验,吸取供试品溶液 15 μL、对照品溶液10 μL、阴性对照品溶液15 μL分别点于同一硅胶 G(市售)薄层板上,以正己烷 -乙酸乙酯 -甲醇(6.4∶3.6∶1)为展开剂,氨蒸气饱和15 min后展开,取出,晾干,喷以稀碘化铋钾试液。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,而阴性对照品溶液色谱无此斑点(图1),表明处方中其他药材对制川乌、制草乌的鉴别无干扰。在制剂有效期内,该鉴别均呈正反应,且3批样品检验结果均满意。
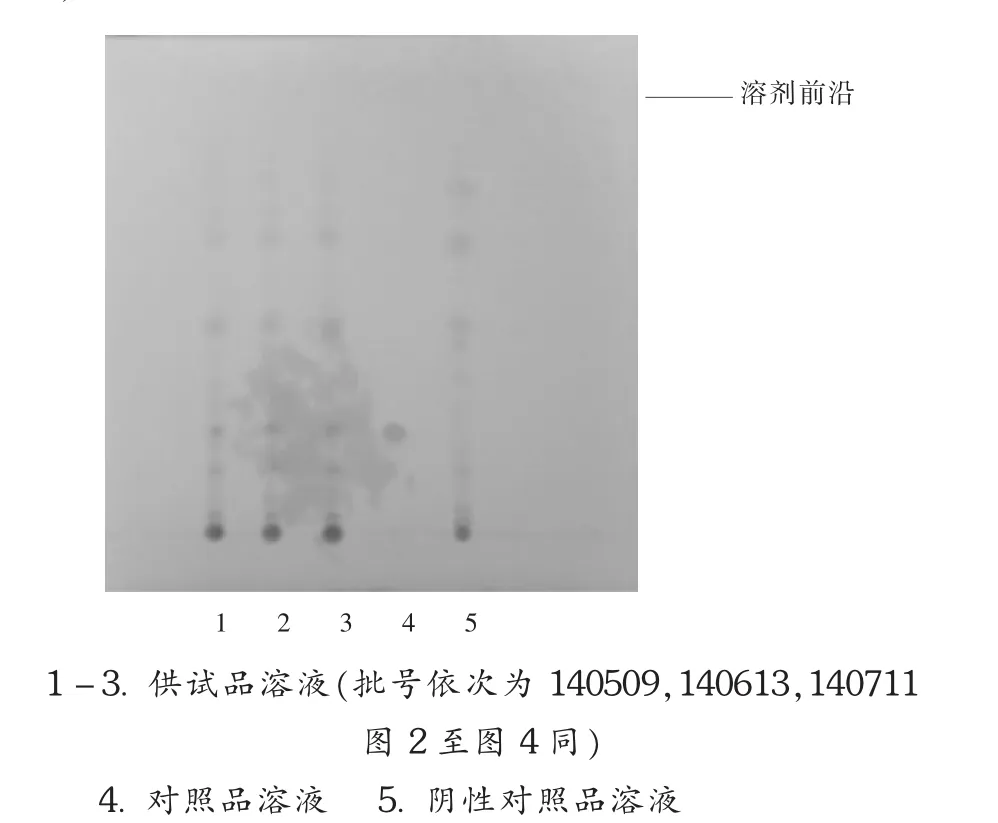
图1 制川乌、制草乌中苯甲酰次乌头原碱鉴别的薄层色谱图
2.1.2 制天南星
按照2010年版《中国药典(一部)》制天南星鉴别项下方法,选择干姜作为观察指标对制剂中的制天南星药材进行薄层鉴别。取本品20 mL,挥去乙醇,加水30 mL使溶解,用乙醚振摇提取3次,每次15 mL,合并乙醚提取液,挥干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取干姜对照药材0.5 g,加乙醇50 mL,加热回流1.5 h,滤过,滤液蒸干,同供试品溶液制备方法制成对照药材溶液。按处方比例称取本制剂中除制天南星和干姜以外的药材,研细,按本品制备工艺及供试品溶液制备方法制成阴性对照品溶液。照薄层色谱法(2010年版《中国药典(一部)》附录Ⅵ B)试验,吸取供试品溶液 15 μL、对照品溶液10 μL、阴性对照品溶液15 μL,分别点于同一硅胶G(市售)薄层板上,以环己烷-乙醚-丙酮-冰醋酸(40 ∶10 ∶5 ∶0.5)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰。供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色斑点,而阴性对照品溶液色谱中无此斑点(图2),表明处方中其他药材对制天南星的鉴别无干扰。在制剂有效期内,该鉴别均呈正反应,且3批样品检验结果均满意。

图2 制天南星鉴别的薄层色谱图
2.1.3 当归
按照2010年版《中国药典(一部)》“当归”鉴别项下方法,选择当归和川芎作为观察指标对制剂中的当归药材进行薄层鉴别。取本品10 mL,挥去乙醇,加水30 mL使溶解,用正丁醇振摇提取3次,每次10 mL,正丁醇提取液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取当归与川芎对照药材各0.5 g,加75%乙醇50 mL,加热回流1 h,滤过,蒸干,残渣加水30 mL使溶解,用正丁醇振摇提取3次,同供试品溶液制备方法分别制成当归对照药材溶液与川芎对照药材溶液。按处方比例称取除当归和川芎以外的药材,研细,按本品制备工艺及供试品溶液制备方法制成阴性对照品溶液。照薄层色谱法(2010年版《中国药典(一部)》附录Ⅵ B)试验,吸取供试品溶液、对照药材溶液、阴性对照品溶液各20 μL,分别点于同一硅胶G(市售)薄层板上,以正己烷-乙酸乙酯(9 ∶1)为展开剂,展开,晾干,在紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色荧光斑点,而阴性对照品溶液色谱中无此斑点(图3),表明处方中其他药材对当归的鉴别无干扰。在制剂有效期内,该鉴别均呈正反应,且3批样品检验结果均满意。
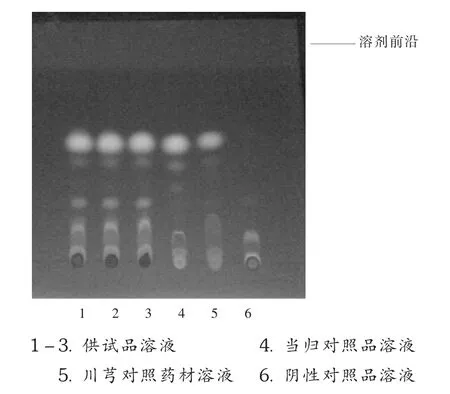
图3 当归鉴别的薄层色谱图
2.2 乌头碱限量测定
乌头碱是存在于生川乌、生草乌等植物中的主要毒性成分,口服纯乌头碱0.2 mg即可中毒。双乌止痛酊处方中虽然使用的是制川乌、制草乌两味中药,但仍存在一定的毒性成分。为进一步确保用药安全,参照2010年版《中国药典(一部)》“成方制剂”及相关文献中关于乌头碱限量的规定[5-6]对双乌止痛酊中的乌头碱进行含量限度检查。取本品40 mL,挥去乙醇,加乙醚-二氯甲烷混合液(3 ∶1)50 mL 溶解,加氨试液 4 mL,密塞,摇匀,放置过夜;振摇1 h,滤过,残渣用乙醚-二氯甲烷混合液(3 ∶1)洗涤 3 次(20,10,10 mL),合并滤液,滤液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取乌头碱对照品,加甲醇制成每1 mL含1.0 mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典(一部)》附录Ⅵ B),吸取供试品溶液 12 μL、对照品溶液2 μL,分别点于同一硅胶G(市售)薄层板上,以正己烷-乙酸乙酯 -甲醇(6.4 ∶3.6 ∶1)为展开剂,氨蒸气饱和15 min后展开,取出,晾干,喷以稀碘化铋钾试液。供试品溶液色谱中,在与对照品溶液色谱相应位置上不出现斑点(图4),符合规定。在制剂有效期内,3批样品检验结果均满意。
2.3 检查
3批样品均符合酊剂项下有关的各项规定(2010年版《中国药典(一部)》附录ⅠN)。
2.4 苯甲酰新乌头原碱含量测定(高效液相色谱法)
2.4.1 色谱条件[6]与系统适用性试验
色谱柱:InertsustainC18柱(250 mm ×4.6 mm,5 μm);流动相:乙腈 - 0.02 mol/L 磷酸二氢钠(25 ∶75);检测波长:230 nm;柱温:30 ℃;流速:1.0 mL /min;进样量:20 μL。理论板数按苯甲酰新乌头原碱峰计算不低于20 000。
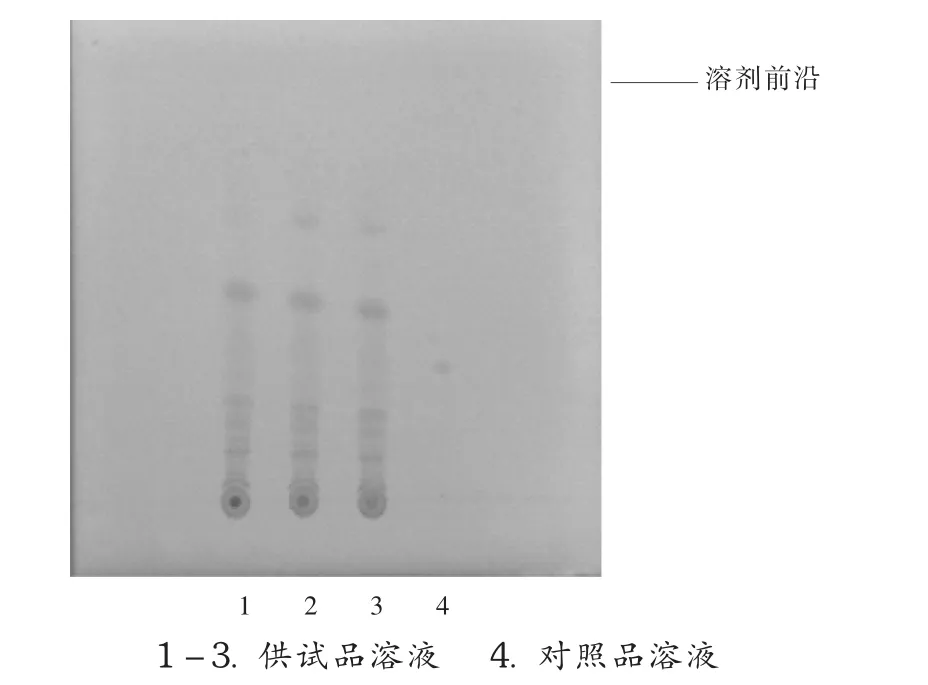
图4 乌头碱限量测定的薄层色谱图
2.4.2 溶液制备
称取苯甲酰新乌头原碱对照品适量,精密称定,置100 mL容量瓶中,加入0.05%盐酸甲醇溶液稀释至刻度,摇匀,配制成含苯甲酰新乌头原碱0.124 4 g/L的对照品贮备液;精密吸取苯甲酰新乌头原碱对照品贮备液4 mL,置10 mL容量瓶中,加0.05%盐酸甲醇稀释至刻度,摇匀,作为对照品溶液。精密吸取本品8 mL,置10 mL容量瓶中,加0.05%盐酸甲醇至刻度,过滤,取续滤液,作为供试品溶液。按处方比例和供试品制备工艺,不加制川乌和制草乌制成阴性样品,按照供试品溶液的制备方法制备阴性对照品溶液。
2.4.3 测定法
精密吸取对照品溶液与供试品溶液各20 μL,分别注入液相色谱仪,记录色谱图。按外标法以峰面积计算,即得。
2.4.4 方法学考察
专属性试验:精密吸取 2.4.2项下 3种溶液各20 μL,按拟订方法测定,记录色谱图,见图5。供试品溶液色谱中,在与对照品溶液色谱相应保留时间处均有相应的色谱峰出现,而阴性对照品溶液则无此峰,表明阴性对照无干扰。
线性关系考察:分别精密吸取对照品贮备液1.0,2.0,4.0,6.0,8.0 mL,置 10 mL 容量瓶中,用0.05% 盐酸甲醇溶液稀释至刻度,按拟订色谱条件进样测定,以对照品质量浓度(X)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程 Y=27 431.8 X-36 696.5,r=1.000 0(n=5)。结果表明,苯甲酰新乌头原碱质量浓度在 0.012 44~0.099 52 g/L范围与峰面积线性关系良好。
精密度试验:取2.4.2项下对照品溶液,按拟订色谱条件连续进样5次,测定峰面积。结果的 RSD为0.96%(n =5),表明仪器精密度良好。
稳定性试验:取同一批(批号为160613)样品,依法制备供试品溶液,常温下分别于 0,1,2,4,6,8 h 时进样,记录峰面积。结果苯甲酰新乌头原碱峰面积在4 h内较稳定;到6 h时开始下降,同时峰形也变差,但 RSD值(2.58%)仍符合规定,表明供试品溶液在6 h内稳定性良好;到8 h时峰面积进一步降低,总结原因,可能跟本方法选择的溶剂有关。苯甲酰新乌头原碱在甲醇中不稳定,而在三氯甲烷或二氯甲烷中较稳定,但前期试验表明,在本研究中的流动相下,采用三氯甲烷作溶剂,供试品溶液色谱峰形较差,故本研究中采用0.05%甲醇作为溶剂。在酸性甲醇中样品的稳定性增加,但仅在6 h内结果稳定。
重复性试验:取同一批(批号为160613)样品5份,依法制备供试品溶液,进样测定。结果的 RSD为1.60%(n=5),表明方法重复性良好。
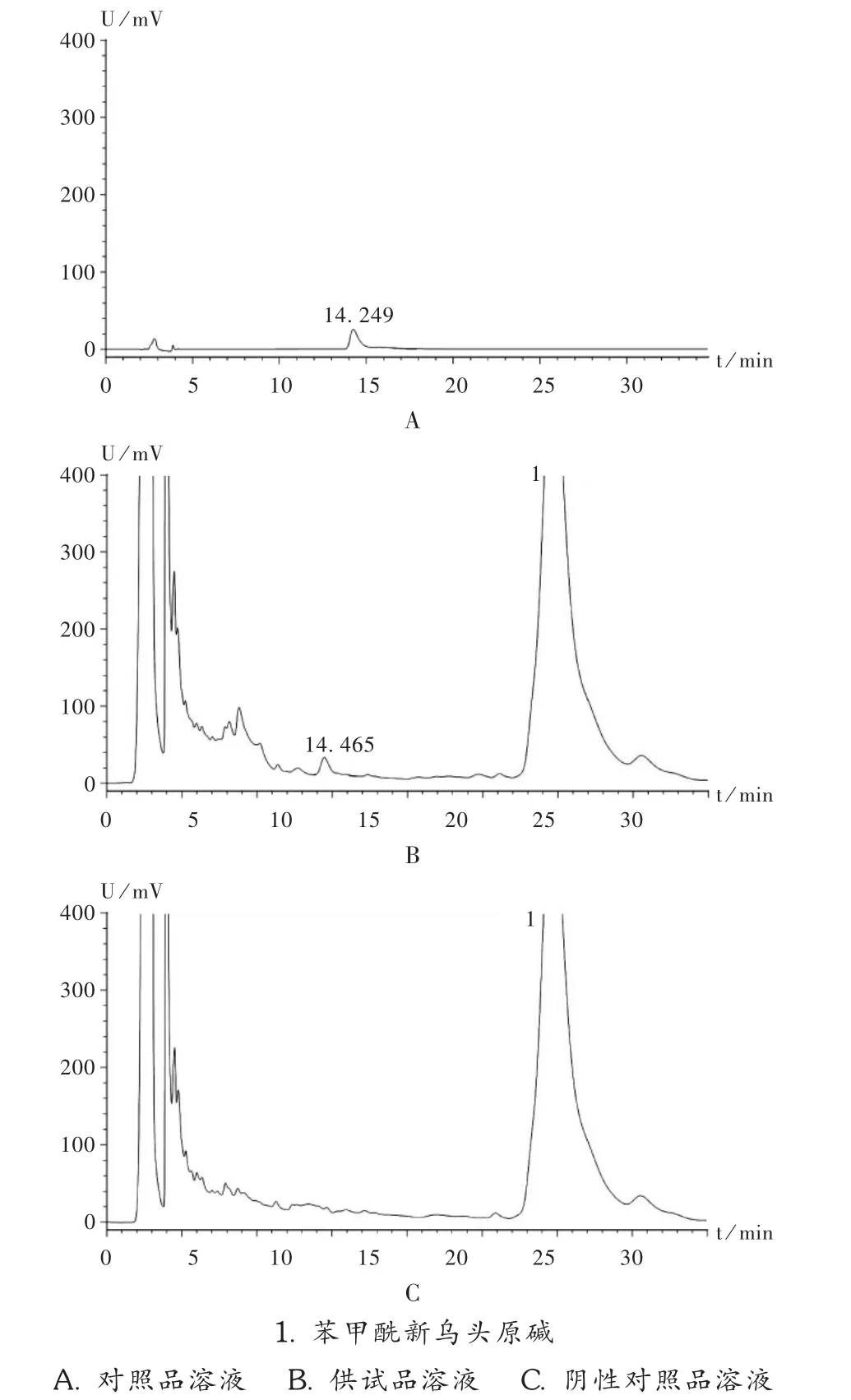
图5 双乌止痛酊高效液相色谱图( =230 nm)
加样回收试验:精密吸取已知含量的样品(批号为160613)4 mL,共 9份,分别置10 mL容量瓶中,分别精密加入对照品贮备液1,2,3 mL,按供试品溶液制备方法制备溶液,精密吸取溶液各20 μL,注入色谱仪,测定含量,计算回收率。结果见表1。
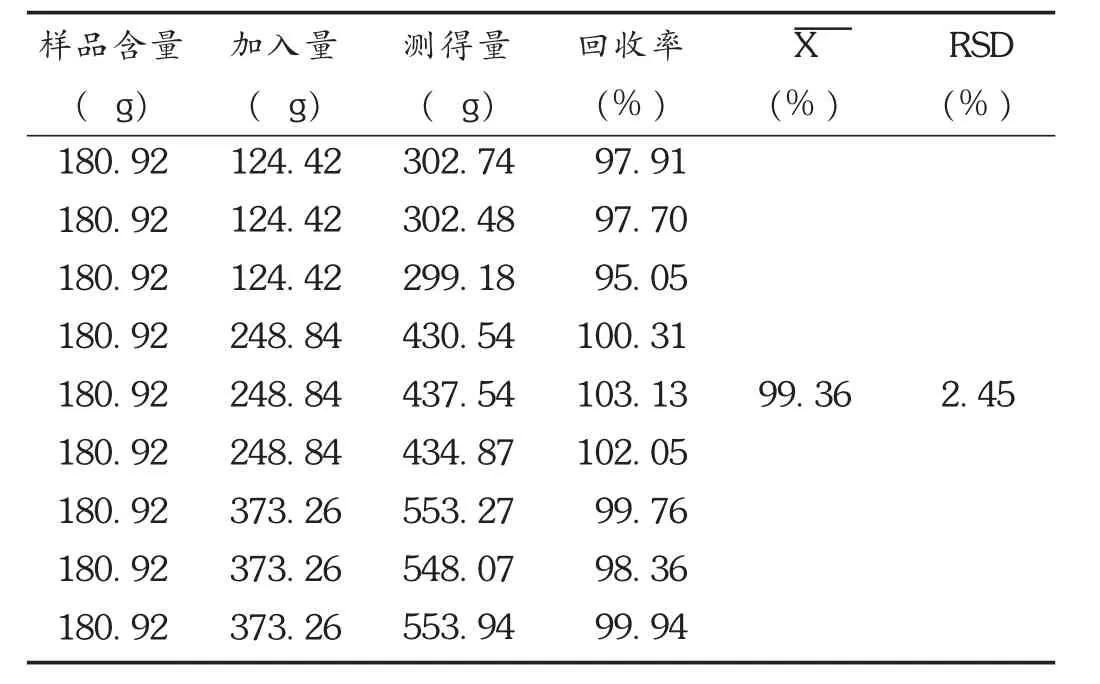
表1 苯甲酰新乌头原碱加样回收试验结果(n=9)
检测限与定量限测定:将对照品溶液逐渐稀释,进样分析,当信噪比(S/N)=3时即为最低检测限,当S/N=10时即为最低定量限。拟订的色谱条件下,苯甲酰新乌头原碱的最低检测限为128 ng,最低定量限为327 ng。
2.4.5 样品含量测定
取3批双乌止痛酊(批号分别为 160509,160613,160711),依法制备供试品溶液,进样测定。结果3批样品中苯甲酰新乌头原碱含量依次为 37.40,45.23,45.30 μg/mL。综合测定结果,最终确定本品每 1 mL 含制川乌、制草乌以苯甲酰新乌头原碱计不得低于20 μg。
3 讨论
3.1 薄层色谱鉴别
双乌止痛酊的有效成分复杂多样,制川乌、制草乌、制天南星等药材中含有的生物碱类成分具有消炎镇痛、抗菌杀病毒的功效[7-8],而其中当归、川芎等药材中含有的阿魏酸、藁本内酯等有效成分可以发挥舒筋活络、祛瘀定痛的功效[9-10]。因此,本研究中选择与制剂疗效相关的制川乌、制草乌以及制天南星、当归作为定性鉴别的重要指标。在制订制川乌、制草乌的薄层色谱标准时,根据前期液相含量测定的结果(苯甲酰新乌头原碱的含量最高,其次是苯甲酰次乌头原碱和苯甲酰乌头原碱),首先选择苯甲酰新乌头原碱为对照品,样品和对照品斑点均很清晰,但可能由于该制剂成分复杂,故制剂阴性位置处存在疑似干扰,重复试验仍存在同样的问题。进一步选择苯甲酰次乌头原碱为对照品,供试品溶液色谱中,与对照品溶液色谱相应保留时间处显相同颜色的斑点,且阴性对照无干扰。因此,本研究中制川乌、制草乌的薄层定性鉴别以苯甲酰次乌头原碱为对照品。
3.2 流动相及检测指标的选择
双乌止痛酊处方中含有制川乌、制草乌以及元胡、干姜、天南星等多味中药,成分十分复杂。其中制川乌、制草乌为方中君药,因此研究之初拟通过测订制川乌及制草乌的有效成分苯甲酰新乌头原碱、苯甲酰乌头原碱和苯甲酰次乌头原碱的含量来控制制剂的质量。首先参照2010年版《中国药典(二部)》中川乌药材中关于苯甲酰新乌头原碱的含量测定方法进行了选择试用:流动相A为乙腈 -四氢呋喃(25 ∶15),流动相 B 为 0.02 mol/L的醋酸铵缓冲液(每1 000 mL加入冰醋酸0.5 mL),梯度洗脱(0~48 min,15% ~26%A;48~48.1 min,26% ~35%A;48.1~58min,35%A;58.1~65min,35% ~15%A)。按照此梯度时,苯甲酰新乌头原碱出峰时间合适,但峰形稍有拖尾,而苯甲酰乌头原碱和苯甲酰次乌头原碱分离度不好,进一步的试验发现,此流动相下制剂中其他成分干扰阴性样品出峰;选择乙腈-0.02 mol/L的磷酸二氢钠(45∶55)为流动相时,发现苯甲酰新乌头原碱、苯甲酰乌头原碱和苯甲酰次乌头原碱出峰时间稍快,且样品峰分离度不好;进一步调整流动相比例为乙腈 -0.02 mol/L 的磷酸二氢钠(25 ∶75),样品峰出峰时间合适,分离度等条件都较好,且后续试验证明在此流动相下阴性样品无干扰,故选择其为最佳流动相。
在上述流动相条件下,对制剂的样品含量进行了初步测定,发现双乌止痛酊中苯甲酰新乌头原碱含量为47 μg/mL,而苯甲酰乌头原碱和苯甲酰次乌头原碱的含量较少,仅分别为 4.72 μg /mL 和 13.76 μg/mL,已接近样品的定量限的下限。考虑到药材本身产地及批次的差异,以及车间大生产的实际,为日后快速准确地检定样品,最终决定选择该制剂中含量较多的苯甲酰新乌头原碱的含量作为质量控制指标。
3.3 检测波长的选择
用紫外分光光度计对苯甲酰新乌头原碱对照品溶液进行全波长扫描,发现苯甲酰新乌头原碱的最大吸收波长为230 nm,故取其为检测波长。
3.4 样品溶剂及提取方法的选择
乌头碱类生物碱结构不稳定,文献报道,纯甲醇、无水乙醇、乙酸乙酯等多种溶剂均能使其结构转化,使检测结果不易重复[2,11-12]。因此,参阅文献[6,13 - 14],比较了常用的异丙醇-三氯甲烷(1∶1)和0.05%盐酸甲醇2种溶剂对样品峰形及稳定性的影响,发现采用前者溶解的供试品峰形较差,并且曲线波动较大,故采用0.05%盐酸甲醇作为溶剂较合适。后续样品的稳定性研究中,发现样品在酸性甲醇中稳定性良好,但仅在6 h内结果稳定。
对于样品的处理方法,前期的含量测定结果显示样品中苯甲酰新乌头原碱含量约为45 μg/mL,含量并不高。故首先考虑将样品在40℃以下低温蒸干,然后用0.05%盐酸甲醇溶解后浓缩定容,作为供试品溶液液。试验结果显示,低温浓缩法处理的样品峰形欠佳,同时由于乌头碱类生物碱的结构特点,导致所测样品中的少量新乌头碱遇热就会水解成苯甲酰新乌头原碱,同时,苯甲酰新乌头原碱在受热的情况下本身也会部分水解。因此,该处理方法所测的含量并不准确。进一步对样品采用简单地直接稀释处理方法,发现该方法下样品峰形较好,曲线也较平稳,同时所测得的含量值更接近理论投料含量。因此,经过比较和试验,本研究中采取将样品稀释作为最终的样品处理方式。该方法简单易操作,同时也避免了湿热可能导致的水解,使样品含量值更稳定和准确。
[1]费建红,宋炳生,陈安兰.双乌止痛酊的质量控制[J].中草药,2002,33(12):1085 - 1086.
[2]张聿梅,鲁 静,蒋 渝,等.川乌和制川乌中单酯及双酯型生物碱成分的含量测定[J].药物分析杂志,2005,25(7):807-811.
[3]Taki M,Niitu K,Omiya Y,et al.8 - O - cinnamoylneoline,a new alkaloid from the flower buds of Aconitum carmichaeli and its toxic and analgesic activities[J].Planta Med,2003,69(9):800 - 803.
[4]陈东安,易进海,黄志芳,等.附子煎煮过程中酯型生物碱含量的动态变化[J].中国实验方剂学杂志,2011,17(3):64 -68.
[5]王兵娥,焦 玉,黄德红.消肿止痛洗剂的薄层色谱鉴别和乌头碱限度检查[J].湖北中医杂志,2013,35(4):72 - 73.
[6]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:220-222,792 -793,附录Ⅵ D.
[7]艾 嫦,朱妍妍,赵长琦.乌头属植物化学成分、药理作用及其内生菌的研究进展[J].天然产物研究与开发,2012,24(2):248-259.
[8]李梦然,曲 玮,梁敬钰.乌头属化学成分和药理作用研究进展[J].海峡药学,2010,22(4):1 - 6.
[9]黄珍辉,盛文兵,刘艳群,等.阿魏酸酯类衍生物的药理作用及合成研究进展[J].中国医药导报,2012,9(19):8 -9.
[10]左爱华,王 莉,肖红斌.藁本内酯药理学和药代动力学研究进展[J].中国中药杂志,2012,37(22):3350 -3353.
[11]李德斌,黄志芳,刘云华,等.炮天雄质量标准研究[J].中国实验方剂学杂志,2013,19(23):146 - 150.
[12]李丽敏,王欣美,王 柯,等.RP-HPLC测定镇痛活络酊中3 种乌头类生物碱含量[J].中成药,2008,30(12):1785.
[13]毛坤军,潘以琳,李富强,等.HPLC同时测定乌七祛风散片中4种生物碱的含量[J].中国实验方剂学杂志,2014,20(22):74 -77.
[14]崔恩忠,唐安福,刘文雅,等.高效液相色谱法测定益肾丸中特女贞苷含量 [J].医学研究生学报,2015,28(11):79 -81.
Quality Standard of Shuangwu Zhitong Tincture
Su Hua, Qian Wenhui,Liao Xin, Bai Ru,Ren Haixiang
(Department of Preparation Division,Nanjing General Hospital of Nanjing Military Command of PLA,Nanjing,Jiangsu,China 210002)
Objective To establish the quality standard of Shuangwu Zhitong Tincture.Methods TLC method was adopted for the qualitative identification ofRhizoma Arisaematis,Angelica Sinensis and benzoylhypaconine in Shuangwu Zhitong Tincture and the limited quantity of aconitine was determined.HPLC method was used to determine the content of benzoylmesaconitine in Shuangwu Zhitong Tincture,the Inertsustain C18column (250 mm × 4.6 mm,5 μm) was adopted,the mobile phase system was acetonitrile-0.02 mol/L NaH2PO4(25 ∶75),the detection wavelength was set at 230 nm,the column temperature was 30 ℃ ,and the flow rate was 1.0 mL /min.Results The qualitative identification ofRhizoma Arisaematis, Angelica Sinensis,and benzoylhypaconine in Shuangwu Zhitong Tincture was specific,the limited quantity of aconitine accorded with the regulations.The liner range of benzoylmesaconitine was 0.012 44 -0.099 52 g/L,r=1.000 0(n =5),and the average recovery was 99.36% ,RSD =2.45% (n =9).Conclusion The method is simple,accurate and reliable,which can be used for the quality control of Shuangwu Zhitong Tincture.
Shuangwu Zhitong Tincture;benzoylmesaconitine;quality standard;TLC;HPLC
R284.1
A
1006-4931(2017)20-0001-05
10.3969 /j.issn.1006 - 4931.2017.20.001
全军医疗机构制剂标准提高科研专项课题重点项目[14ZJZ08]。
苏华(1978-),女,硕士研究生,主管药师,研究方向为医院制剂学和靶向制剂学,(电子信箱)suhuash@sina.com。
任海祥(1966-),男,硕士研究生,副主任药师,研究方向为医院制剂学,(电话)025-80860166(电子信箱)rhx1111@sina.cn。
2017-07-10)
