酱香型白酒第二轮次酒发酵过程微生物多样性研究
2017-10-24黄蕴利黄永光胡建峰钟方达
黄蕴利,黄永光*,胡建峰,胡 峰,钟方达
(1.贵州大学 酿酒与食品工程学院,贵州 贵阳 550025;2.贵州大学 贵州省发酵工程与生物制药重点实验室,贵州 贵阳 550025;3.贵州茅台酒厂(集团)习酒有限责任公司,贵州 习水 564622)
酱香型白酒第二轮次酒发酵过程微生物多样性研究
黄蕴利1,2,黄永光1,2*,胡建峰3,胡 峰3,钟方达3
(1.贵州大学 酿酒与食品工程学院,贵州 贵阳 550025;2.贵州大学 贵州省发酵工程与生物制药重点实验室,贵州 贵阳 550025;3.贵州茅台酒厂(集团)习酒有限责任公司,贵州 习水 564622)
为探究酱香型白酒二轮次发酵过程中微生物群落结构的变化、微生物多样性以及物种与样品之间的关系,应用高通量测序技术对酱香型白酒第二轮次酒生产堆积及窖池发酵过程酒醅中微生物的多样性及其主要功能菌群进行研究。结果表明,在堆积过程中共检测到细菌138个属,真菌54个属;窖池发酵过程中共检测到细菌262个属,真菌267个属。酒醅中主要细菌类群为Firmicutes和Proteobacteria,主要真菌类群为Ascomycota和Basidiomycota。堆积过程中绝对优势细菌属有Bacillus、Enterococcus、Lactococcus、Lactobacillus;绝对优势真菌属有Thermoascus、Thermomyces、Candida、Aspergillus。在窖池发酵酒醅中其绝对优势细菌属为Lactobacillus;绝对优势真菌属为Saccharomyces、Candida、Penicillium、Fusarium。由于堆积、窖池发酵酒醅所处发酵环境、发酵物料物态及其工艺参数差异较大原因,使得两者之间生物物种存在较大差异。
酱香型白酒;高通量测序;酒醅;微生物群落多样性
中国白酒酿造工艺是固态条件下独特而复杂的自发发酵过程[1-2]。中国白酒类是使用谷物(如高粱、小麦和大麦)进行同步糖化发酵(simultaneous saccharification and fermentation,SSF)的过程。同步糖化发酵结合酶解淀粉为还原糖,为微生物提供大量的碳源[3]。此外,白酒发酵过程含有多种微生物,包括酵母、细菌和丝状真菌[4]。复杂的丝状真菌群落产生多种水解酶降解淀粉为多糖,包括葡萄糖、半乳糖、麦芽糖和蜜二糖[5]。由于微生物的代谢活动和固体培养基的低导热性使发酵剂温度能达到50℃的高温[6]。此外,在酒精发酵阶段有两个因素影响微生物的生长,一是低的pH值(pH3.0),二是产生的乙醇(4.5%vol~5.5%vol)抑制微生物的生长[7]。这样的恶劣同步糖化发酵(SSF)环境导致具有特定的生理特性和性能的微生物得到富集[8],特别是具有独特的耐热和耐酸性能的微生物[9]。第二轮次酒能够增加基酒中酒体的曲香味,能够使糟醅中累积更多的微生物代谢产物,有利于基酒香味物质的积累,在后期轮次相应形成更多的酱香型酒前驱物质,同时为后期轮次多产酱香奠定基础。
在20世纪80~90年代,许多关于酒醅微生物的研究主要是通过传统的可培养方法进行的[10]。然后在接下来的十年中,开始采用不可培养方法对酒醅中的微生物群落进行研究[11-12]。然而,这些文献报道的主要是通过构建16S rRNA基因或变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)的克隆文库进行研究。DGGE的局限性存在一个条带内可能含有多个物种,很难清晰鉴别[13]。16SrRNA基因克隆文库的构建,属于劳动密集型方法,只允许分析有限数量的16S rRNA基因序列[14]。由于这些常规分子生物学技术的固有局限性,应用这些方法去认识酒醅中复杂的微生物生态还远远不够。因此,需要采用更科学系统的方法才能全面了解酒醅发酵过程的微生物多样性和主要功能群落的分布。高通量测序技术(high-throughputsequencing,HTS)的最新进展使得能够通过对许多样品进行平行的深入分析,以更低的成本和更易加工来显著提高生产率。16SrRNA区域的扩增与高通量多序列读取的产生能够获得覆盖完全的微生物群落[15]。目前,这种新兴技术已被用于研究不同发酵食品中的微生物生态,包括醋[16]和牛奶[17]以及浓香型白酒[18]和清香型白酒中[19-20]。郭敏等[21]也证实了高通量测序技术可用于酱香型白酒酿造发酵过程酒醅微生物多样性的研究。
本研究旨在通过Illumina MiSeq平台研究分析酱香型白酒第二轮次发酵过程细菌16S rRNA和真菌内转录间隔区(internal transcribed spacer,ITS)高变区,研究生产堆积、窖池发酵酒醅中的微生物多样性及其主要功能群落结构,深入认识发酵过程的微生物的多样性及其所形成的发酵机理,为传统白酒酿造、固态发酵及食品发酵工业提供参考。
1 材料与方法
1.1 材料与试剂
酒醅:取自XJ酱香型白酒第二轮次酒生产堆积、窖池发酵酒醅,共25个酒醅样品;DNA提取试剂盒:美国Omega Bio-Tek公司。
1.2 仪器与设备
SW-CJ-1F型超净工作台:苏州净化设备有限公司;Heraeus Multifuge X3高速冷冻离心机:美国赛默飞世尔科技公司;HQ-60-Ⅱ漩涡混合器:北京北方同正生物技术发展有限公司;HH-4数显恒温水浴锅:国华电器有限公司;ABIGeneAmpR9700型聚合酶链反应(polymerasechainreaction,PCR)仪:美国ABI公司;IlluminaMiseq 测序平台:上海美吉生物医药科技有限公司。
1.3 方法
1.3.1 取样方法
堆积过程酒醅的取样周期为1 d,收堆时计为0 d,取样点如图1所示,即上层A点(代表堆子上部一个层面);中层B、C两点取样混匀后作为一个样品(代表堆子中部层面);下层D、E两点取样混匀后作为一个样品(代表堆子下部层面)。窖池发酵过程酒醅取样周期为5 d,入池时计为0 d,以后每隔5 d取样一次,同一层面分别确定三个取样点(见图2);将同一层面的A、B、C三点所取酒醅混匀为一个样品,D、E、F三点所取酒醅混匀为一个样品,G、H、I三点的取酒醅混匀为一个样品,分别代表窖池中上、中、下三个层面的酒醅样品。
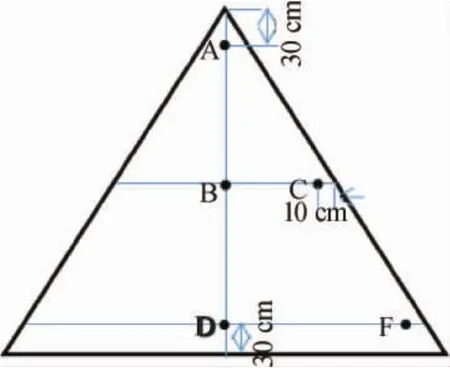
图1 堆积酒醅取样点Fig.1 Sampling points of fermented grains in the accumulation process
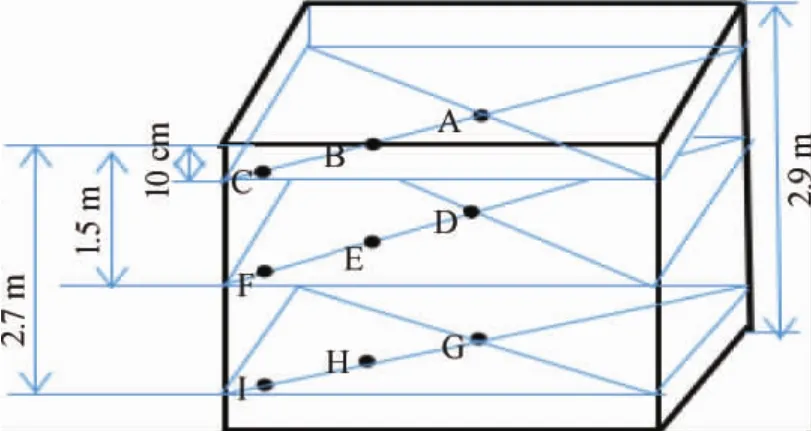
图2 窖池发酵酒醅取样点Fig.2 Sampling points of fermented grains in the fermentation process
1.3.2 样品预处理及DNA提取
分别称取每个样品10 g,用15 mL灭菌后的0.1 mol/L磷酸盐缓冲液(phosphate buffer solution,PBS)悬浮,加入三颗玻璃珠,漩涡振荡10 min。300 r/min离心5 min,取上清,沉淀用PBS缓冲液重复洗涤3次,离心后收集上清,混匀所收集上清。将上清于12 000 r/min离心5 min,弃上清,收集细胞沉淀。用5 mL PBS缓冲液洗3次沉淀,每次于12 000 r/min离心5 min后去上清,收集并混匀沉淀[22]。
应用土壤总DNA提取试剂盒,根据说明书提示,进行堆积、窖池发酵酒醅样品中提取的微生物细胞的总DNA提取。
1.3.3 16S rRNA、ITS基因扩增
细菌使用引物338F(5'-ACTCCTACGGGAGGCAGCA G-3')和806R(5'-GGACTACHV GGGTWTCTAAT-3')扩增V4高变区,真菌使用引物ITS1F(5'-CTTGGTCATTTAGAGG AAGTAA-3')和2043R(5'-GCTGCGTTCTTCATCGATGC-3')扩增高变区。20μLPCR混合体系中含FastPfu聚合酶0.4μL,前引物(5 μmol/L)0.8 μL,后引物(5 mmol/L)0.8 μL,5×FastPfu缓冲液4 μL,2.5 mmol/L脱氧核糖核苷三磷酸(de-oxy-ribonucleoside triphosphate,dNTP)2 μL和10 ng模版脱氧核糖核酸(DNA)。PCR条件为:95℃预变性3 min;95℃变性30 s;55 ℃复性30 s;72 ℃延伸45 s;共35个循环,最后72℃延伸10 min。
1.3.4 Illumina Miseq测序
将PCR产物用QuantiFluorTM-ST蓝色荧光定量系统进行检测,再按照每个样品的测序量要求,进行相应比例的混合。再应用Illumina Miseq测序平台进行测定分析。
2 结果与分析
2.1 序列数据优化与统计
利用Illumina Miseq测序平台得到样品双端序列数据,在进行reads拼接以及对reads的质量和拼接效果进行质控过滤后,从25个样品中分别获得细菌、真菌的有效序列分别为874 809条和793 524条。
2.2 基于OTU的聚类分析
2.2.1 Alpha多样性分析
通过USEARCH将标签分组成具有97%相似性的个分类单位(operational taxonomic units,OTU)。计算Chao和ACE指数(以确定物种丰富度),Shannon和Simpson指数(以确定物种多样性)以及检测到的物种数目,以描述堆积、窖池发酵酒醅样品中细菌、真菌群落的多样性。
(1)稀疏曲线分析
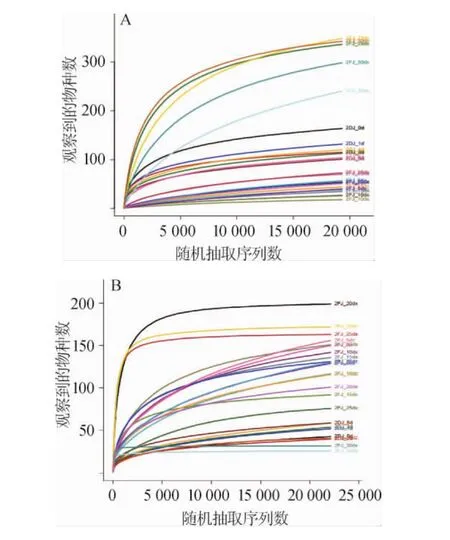
图3 各酒醅样品细菌(A)及真菌(B)的稀疏曲线Fig.3 Dilution curves of bacteria(A)and fungi(B)in different fermented grains samples
稀疏性曲线是从样本中随机抽取一定数量的个体,统计这些个体所代表的物种数目,并以个体数与物种数来构建曲线。它可以用来比较测序数据量不同的样本中物种的丰富度,也可以用来说明样本的测序数据量是否合理。25个酒醅样品中细菌、真菌的稀疏曲线(rarefaction curve)见图3。
由图3A可知,25个酒醅样品细菌的稀释曲线都趋向平坦,说明各样本细菌的测序数据量合理,测序深度已足够。由图3B可知,25个酒醅样品真菌的稀释曲线趋向平坦,说明各样品真菌的测序数据量合理,测序深度已足够。
(2)Shannon-Wiener曲线分析
Shannon-Wiener是反映样本中微生物多样性的指数,利用各样本的测序量在不同测序深度时的微生物多样性指数构建曲线,以此反映各样本在不同测序数量时的微生物多样性。25个酒醅样品细菌、真菌的Shannon-Wiener曲线见图4。
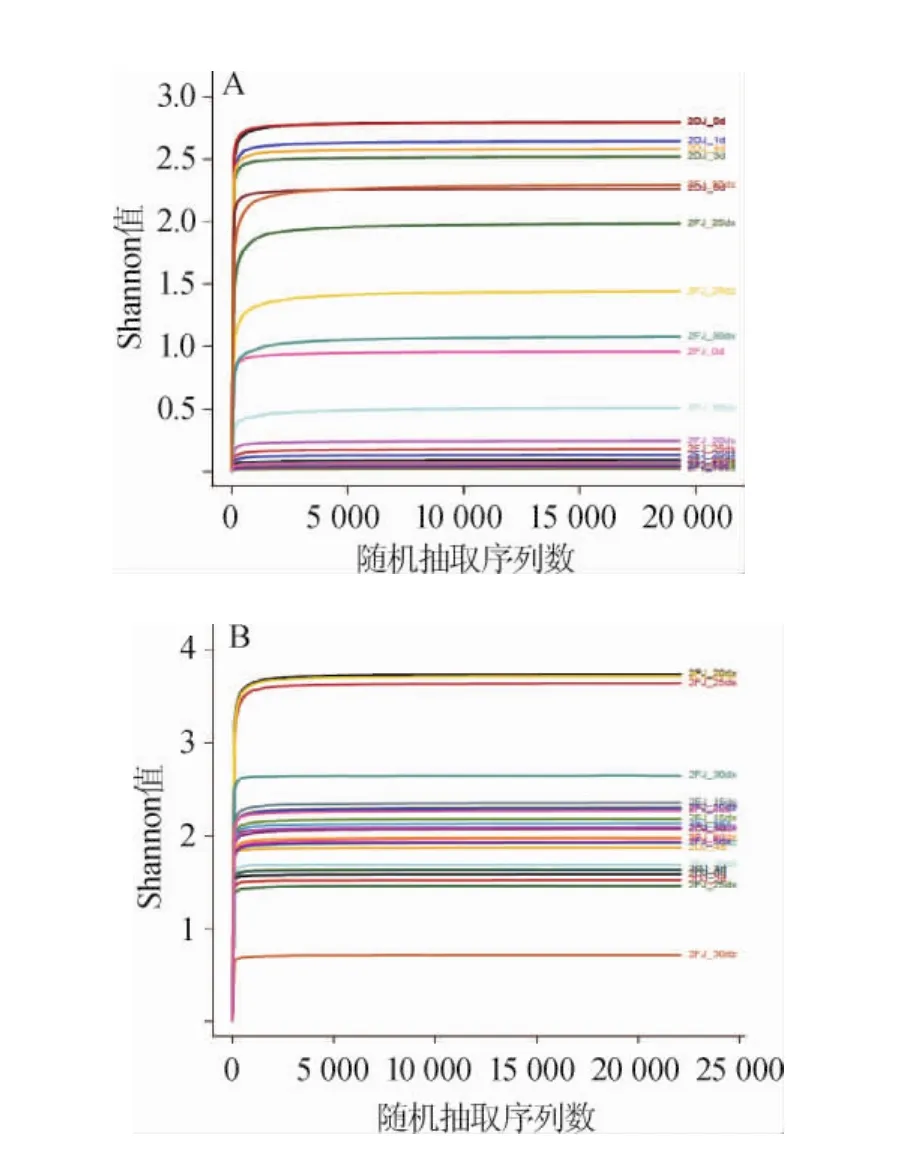
图4 各酒醅样品细菌(A)及真菌(B)的Shannon-Wiener曲线Fig.4 Shannon-Wiener curves of bacteria(A)and fungi(B)in different fermented grains samples
由图4A可知,25个酒醅样品细菌的Shannon-Wiener曲线都趋向平坦,说明测序的数据量足够大,对酒醅中微生物多样性分析基本覆盖酒醅中细菌的种类。由图4B可知,25个酒醅样品真菌的Shannon-Wiener曲线趋向平坦,说明测序的数据量足够大,对酒醅中微生物多样性分析基本覆盖酒醅中真菌的种类。
2.2.2 OTU分布Venn图分析
堆积和窖池上、中、下层发酵酒醅样品的细菌、真菌群落多样性结构OTU分布Venn图见图5。
由图5A可知,四组样品共有的OTU有153个,分别占堆积、窖池发酵上、中、下三层酒醅样品OTU总数的63.75%、37.05%、36.60%和38.06%。窖池发酵上、中、下三层酒醅样品所共有的OTU为347,分别占窖池发酵上、中、下三层酒醅样品OTU总数的84.02%、83.01%和86.32%;堆积和窖池发酵共有的OTU相对较少,而窖池发酵上、中、下三层共有的OTU相对较多,说明堆积和窖池发酵过程酒醅中细菌菌群多样性相差较大,而窖池内发酵酒醅的菌群多样性相差不大。
由图5B可知,四组酒醅样品共有的OTU为55,四组酒醅样品的OTU分别占堆积、窖池发酵上、中、下三层酒醅样品OTU总数的48.25%、12.97%、13.75%和12.53%。窖池发酵上、中、下三层酒醅样品所共有的OTU为179,上、中、下三层酒醅样品的OTU分别占窖池发酵上、中、下三层酒醅样品OTU总数的42.22%、44.75%和40.77%;说明大多数真菌是在窖池发酵过程中产生或相对数量呈现增加,且窖池上、中、下三层酒醅样品的真菌也存在较大差异。
2.3 微生物群落多样性结构分析
堆积和窖池发酵酒醅样品细菌、真菌群落结构在门分类、属分类水平上的结果分别见图6和图7。
由图6可知,酒醅样品中的细菌群落结构组成情况,对堆积和窖池发酵上、中、下三层酒醅样品中主要细菌类群(相对丰度>1%)统计结果见表1。
由表1可知,堆积和窖池发酵酒醅样品中的优势细菌门分类比较明显,均为Firmicutes。在堆积、窖池发酵酒醅样品中,Firmicutes相对丰度均占92%以上;堆积酒醅中优势细菌属有Bacillus(29.22%),Enterococcus(15.46%),Lactococcus(12.82%),Lactobacillus(10.31%),Lentibacillus(8.99%),Kroppenstedtia(6.13%),Enterobacteriaceae(4.8%),Alkaliphilus(2.59%),Oceanobacillus(1.44%),Thermoactinomyces(1.37%),而窖池发酵上层酒醅中存在的主要细菌属类有Lactobacillus(88.86%)、Acetobacter(3.12%)、Bacillus(1.97%),窖池发酵中层酒醅中的主要细菌属类有Lactobacillus(93.29%)、Bacillus(1.48%),窖池发酵下层酒醅中的主要细菌属类有Lactobacillus(95.21%)。
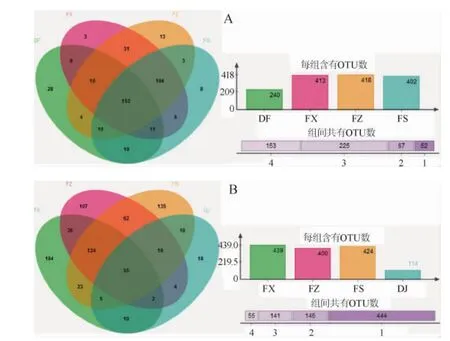
图5 各酒醅样本细菌(A)及真菌(B)的OTU分布Venn图Fig.5 Venn diagram of OTU distribution of bacteria(A)and fungi(B)in different fermented grains samples
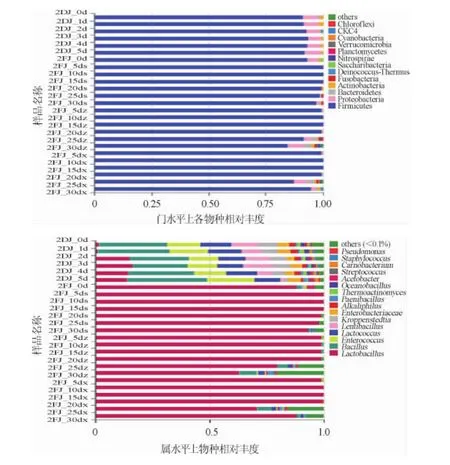
图6 门水平(A)及属水平(B)细菌群落结构图Fig.6 Structure diagram of bacterial community of phylum level(A)and genus level(B)

表1 堆积和窖池发酵酒醅样品中的主要细菌类群Table 1 Main bacterial groups in fermented grains samples of accumulation and pits fermentation process

图7 门水平(A)及属水平(B)真菌群落结构图Fig.7 Structure diagram of fungi community of phylum level(A)and genus level(B)
由图7可知,样品中的真菌群落结构组成情况,对堆积和窖池发酵上、中、下三层酒醅样品中的主要真菌类群(相对丰度>1%)进行统计,结果见表2。
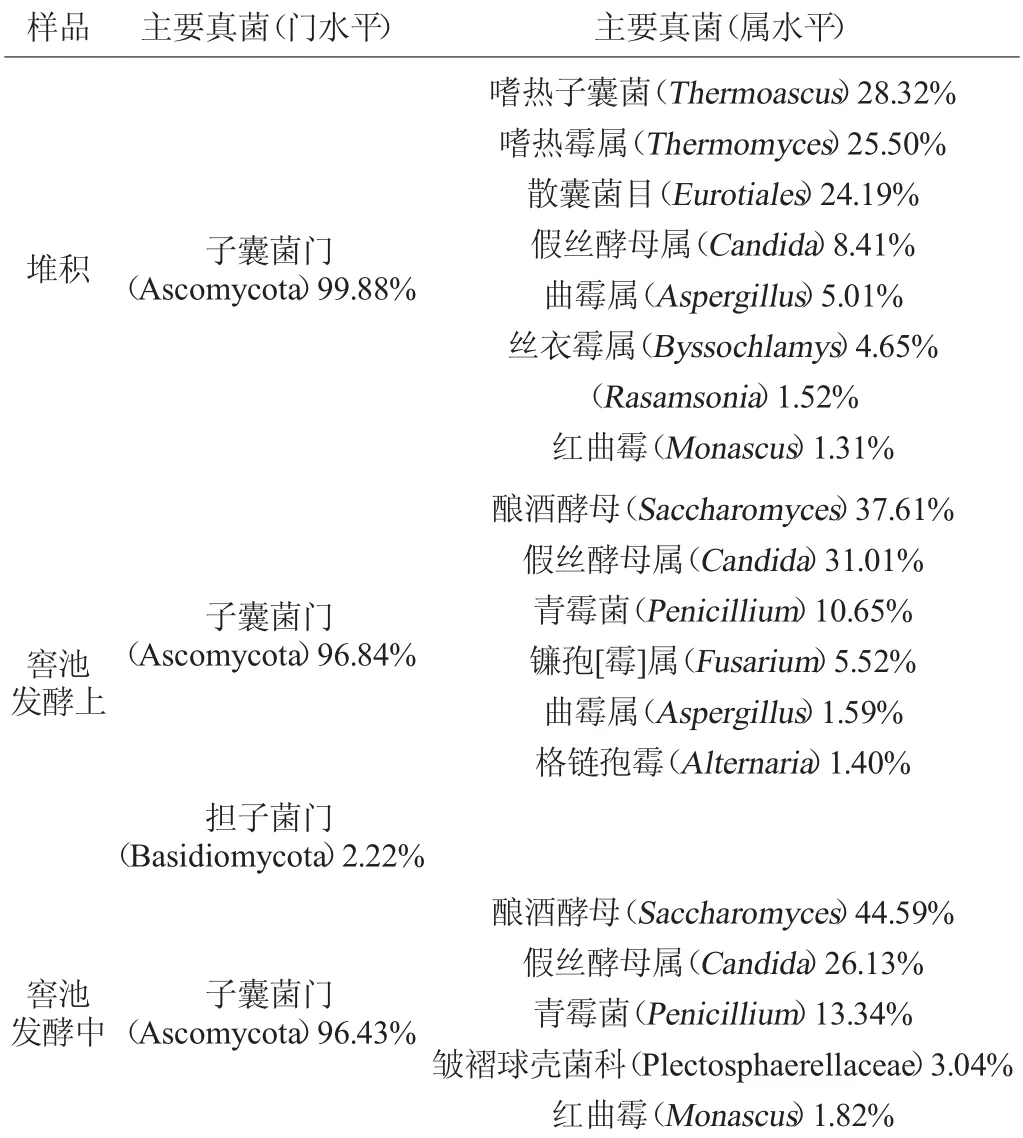
表2 堆积和窖池发酵酒醅样品中的主要真菌类群Table 2 Main fungi groups in fermented grains samples of accumulation and pits fermentation process
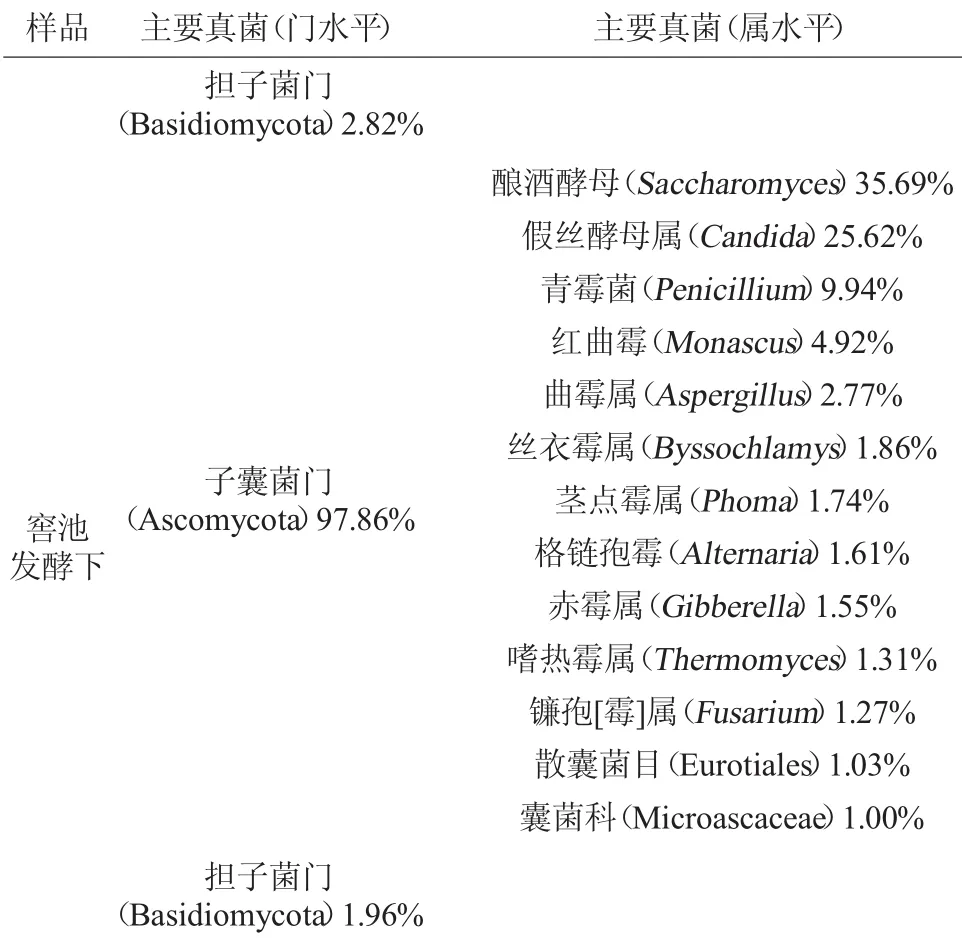
续表
由表2可知,堆积和窖池发酵酒醅样品中的优势真菌门类比较明显,均为Ascomycota。在堆积和窖池发酵酒醅样品中,Ascomycota相对丰度均在96%以上;堆积过程中优势真菌属有Thermoascus(28.32%),Thermomyces(25.50%),Eurotiales(24.19%),Candida(8.41%),Aspergillus(5.01%),Byssochlamys(4.65%),Rasamsonia(1.52%),Monascus(1.31%);而窖池发酵上层酒醅中的主要真菌属类则为Saccharomyces(37.61%),Candida(31.01%),Penicillium(10.65%),Fusarium(5.52%),Aspergillus(1.59%),Alternaria(1.40%);窖池发酵中层酒醅中的主要真菌属类为Saccharomyces(44.59%),Candida(26.13%),Penicillium(13.34%),Plectosphaerellaceae(3.04%),Monascus(1.82%);窖池发酵下层酒醅中的主要真菌属类为Saccharomyces(35.69%),Candida(25.62%),Penicillium(9.94%),Monascus(4.92%),Aspergillus(2.77%),Byssochlamys(1.86%),Phoma(1.74%),Alternaria(1.61%),Gibberella(1.55%),Thermomyces(1.31%),Fusarium(1.27%),Eurotiales(1.03%),Microascaceae(1.00%)。
3 结论
堆积和窖池发酵过程酒醅中细菌多样性相差较大,而窖池内上、中、下三层发酵酒醅由于均处于厌氧发酵条件,其菌群多样性相差不大。大多数真菌是在窖池发酵过程中产生或其相对丰度呈现增加而被检测到,且真菌在窖池上、中、下三层酒醅中也存在较大的差异。
对比酱香型白酒第二轮轮次酒生产堆积、窖池发酵酒醅中微生物多样性,结果表明,二者存在较大的差异,堆积过程中主要细菌(相对丰度>10%)有Bacillus、Enterococcus、Lactococcus、Lactobacillus,而窖池发酵过程中主要为Lactobacillus,其相对丰度>80%;堆积过程中主要真菌(相对丰度>10%)有Thermoascus、Thermomyces、Eurotiales,而窖池发酵过程中主要为Saccharomyces、Candida、Penicillium。主要是由于堆积、窖池发酵酒醅所处发酵环境、发酵物料物态及其工艺参数差异较大的缘故,堆积处于开放式的好氧、兼氧环境条件下,可富集环境空气中的微生物,且可与空气接触,有较丰富的氧浓度参与堆积发酵,而窖池发酵过程处于绝对厌氧条件,微生物的多样性会随发酵环境条件的变化而发生自控调节,部分微生物的生长会受到抑制,且随着发酵的进行,酒精、酸类等不断积累,也会抑制微生物的生长,自然窖池酒醅中的微生物多样性与堆积酒醅中的微生物多样性出现明显差异。
[1]ZHUS,LUX,JIK,et al.Characterization of flavor compounds in Chinese liquor Moutai by comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry[J].Anal Chim Acta,2007,597(2):340-348.
[2]韩兴林,张五九,李 红.从标准简单分析白酒香型的发展[J].中国酿造,2015,34(5):1-6.
[3]BALLESTEROS M,OLIVA J M,NEGRO M J,et al.Ethanol production from paper material using a simultaneous saccharification and fermentation system in a fed-batch basis[J].World J Microbiol Biotechnol,2002,39(12):1843-1848.
[4]LI X R,MA E B,YAN L Z,et al.Bacterial and fungal diversity in the starter production process of Fen liquor,a traditional Chinese liquor[J].J Microbiol,2013,51(4):430-438.
[5]陈 笔.酱香型白酒酿造过程中霉菌群落结构以及霉菌与酵母相互作用的研究[D].无锡:江南大学,2014.
[6]GLASSEY I,WARD A C.Solid state fermentation[M].New York:Springer,2015,217-225.
[7]徐 岩.基于风味导向技术的中国白酒微生物及其代谢调控研究[J].酿酒科技,2015(2):1-11,16.
[8]WANG C L,SHI D J,GONG G L.Microorganisms in Daqu:a starter culture of Chinese Maotai-flavor liquor[J].World J Microbiol Biotechnol,2008,24(10):2183-2190.
[9]XING MENG,QUN WU,LI WANG,et al.Improving flavor metabolism ofSaccharomyces cerevisiaeby mixed culture withBacillus licheniformis for Chinese Maotai-flavor liquor making[J].J Ind Microbiol Biotechnol,2015,42(10):1601-1608.
[10]赵 爽,杨春霞,窦 屾,等.白酒生产中酿酒微生物研究进展[J].中国酿造,2012,31(4):5-10.
[11]SUN W N,XIAO H Z,PENG Q,et al.Analysis of bacterial diversity of Chinese Luzhou-flavor liquor brewed in different seasons by Illumina Miseq sequencing[J].Ann Microbiol,2016,66(3):1293-1301.
[12]LI K,ZHANG Q,ZHONG X T,et al.Microbial diversity and succession in the Chinese Luzhou-flavor Liquor fermenting cover lees as evaluated by SSU rRNA profiles[J].Indian Microbiol,2013,53(4):425-431.
[13]LIN Y,TONG Z.Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 pyrosequencing[J].Appl Microbiol Biotechnol,2013,97(10):2681-2690.
[14]SUCHODOLSKI J S,DOWD S E,WILKE V,et al.16S rRNA gene pyrosequencing reveals bacterial dysbiosis in the duodenum of dogs with idiopathic inflammatory bowel disease[J].PLoS One,2012,7(6):e39333.
[15]POLKA J,REBECCHI A,PISACANE V,et al.Bacterial diversity in typical Italian salami at different ripening stages as revealed by highthroughput sequencing of 16S rRNA amplicons[J].J Food Microbiol,2015,46(1):342-356.
[16]NIE Z,ZHENG Y,WANG M,et al.Exploring microbial succession and diversity during solid-state fermentation of Tianjin duliu mature vinegar[J].Bioresour Technol,2013,148(8):325-333.
[17]BOKULICH NA,AMIRANASHVILI L,CHITCHYAN K,et al.Microbial biogeography of the transnational fermented milk matsoni[J].Food Microbiol,2015,50(1):12-19.
[18]程 伟,吴丽华,徐亚磊,等.浓香型白酒酿造微生物研究进展[J].中国酿造,2014,33(3):1-4.
[19]张双燕,廖永红,纪 南,等.基于高通量测序技术分析北京清香型大曲微生物多样性[J].中国酿造,2016,35(11):49-53.
[20]雷振河.采用高通量测序技术分析清香型白酒酿造微生物[J].食品与发酵工业,2015,41(9):164-167.
[21]郭 敏,黄永光,邱树毅,等.高通量测序在酱香白酒微生态多样性研究中的应用[J].中国酿造,2017,36(5):146-151.
[22]王海燕.PCR-DGGE技术对清香型汾酒微生物群落结构演变规律的研究[D].无锡:江南大学,2014.
Microbial diversity of the second rounds liquid of Moutai-flavorBaijiuduring fermentation process
HUANG Yunli1,2,HUANG Yongguang1,2*,HU Jianfeng3,HU Feng3,ZHONG Fangda3
(1.School of Liquor and Food Engineering,Guizhou University,Guiyang 550025,China;2.Key Laboratory of Fermentation Engineering and Biological Pharmacy of Guizhou Province,Guizhou University,Guiyang 550025,China;3.Guizhou Moutai Brewery(Group)Xijiu Co.,Ltd.,Xishui 564622,China)
In order to explore the changes of microbial community structure,microbial diversity and the relationship between species and samples in the second rounds liquid of Moutai-flavorBaijiuduring the fermentation process,the microbial diversity and its main functional flora in the fermented grains of Moutai-flavorBaijiusecond rounds liquid accumulation and pits fermentation process were researched by high-throughput sequencing technology.The results showed that the 138 genera of bacteria and 54 genera of fungi were detected in the accumulation process,and the 262 genera of bacteria and 267 genera of fungi were detected in the pits fermentation process.The main bacterial groups in the fermented grains were Firmicutes and Proteobacteria,and the main fungi groups were Ascomycota and Basidiomycota.In the accumulation process,the absolute dominant bacteria wereBacillus,Enterococcus,LactococcusandLactobacillus,and the absolute dominant fungi wereThermoascus,Thermomyces,CandidaandAspergillus.In the pits fermentation process,the absolute dominant bacteria wasLactobacillus,the absolute dominant fungi wereSaccharomyces,Candida,PenicilliumandFusarium.Due to the great difference of fermented grains in accumulation,fermentation environment,fermentation material state,process parameters and pits fermentation process,there were significant differences in biological species.
Moutai-flavorBaijiu;high-throughput sequencing;fermented grains;microbial community diversity
TS261.6
0254-5071(2017)09-0030-06
10.11882/j.issn.0254-5071.2017.09.007
2017-03-07
贵州省科学技术厅重大专项(黔科合重大专项字[2015]6012);贵州省工业攻关项目(黔科合GZ字(2011)3015);贵州省科学技术厅重大专项(黔科合重大专项字[2012]601-5)
黄蕴利(1991-),女,硕士研究生,研究方向为酱香型白酒微生物。
*通讯作者:黄永光(1976-),男,研究员,博士,研究方向为酱香型白酒。
